 2021, Vol. 61
2021, Vol. 61中国科学院微生物研究所,中国微生物学会,中国菌物学会
文章信息
- 辜杨, 项兴本, 王少磊, 曹荣升, 任聪, 范文来, 徐岩. 2021
- Yang Gu, Xingben Xiang, Shaolei Wang, Rongsheng Cao, Cong Ren, Wenlai Fan, Yan Xu. 2021
- 基于窖泥微生物相对定量与绝对定量联用的窖泥老熟特征分析
- Profiling the community structure of pit clay during aging process with the combination of relative and absolute quantitative approaches
- 微生物学报, 61(11): 3444-3457
- Acta Microbiologica Sinica, 61(11): 3444-3457
-
文章历史
- 收稿日期:2021-01-05
- 修回日期:2021-03-14
- 网络出版日期:2021-04-01


引用本文 |



2. 安徽迎驾贡酒股份有限公司, 中国生态白酒研究院, 安徽 六安 237271;
3. 江南大学工业生物技术教育部重点实验, 江苏 无锡 214122
2. Anhui Yingjiagong Distillery Co. LTD, China Ecological Baijiu Research Center, Liu'an 237271, Anhui Province, China;
3. Key Laboratory of Industrial Biotechnology of Ministry of Education, Jiangnan University, Wuxi 214122, Jiangsu Province, China
浓香型白酒酿造采用泥窖固态发酵、续糟配料、混蒸混烧的酿造工艺[1],长期的生产实践经验表明“千年老窖万年糟,酒好全凭窖池老”,窖池作为浓香型白酒发酵的容器,浓香型白酒质量随着窖池窖龄的增加而提高。而老窖产好酒的原因在于窖泥中的厌氧菌群在长期发酵过程中不断驯化富集,形成稳定的具有较高生物与功能多样性的窖泥微生物生态系统[2],在窖泥微生物的作用下,大分子有机物被分解成各类代谢产物,代谢产物之间相互转化形成了浓香型白酒风味物质[3-4]。因此解析窖泥老熟过程中的微生物组成变化与作用,对提高白酒生产质量具有重要意义。
近年来,研究人员基于高通量测序技术对不同老熟程度窖泥内的窖泥菌群组成展开了一系列的研究。随着窖泥老熟,窖泥菌群结构逐步趋于稳定,窖泥内乳杆菌属相对丰度降低,己酸菌属、甲烷杆菌属、甲烷短杆菌属和互营单胞菌属等变为主要微生物[5-7]。目前窖泥菌群组成研究均基于相对丰度进行比较分析,缺少对生物量的校正,导致在观测群落中种群变化时可能出现偏差,尤其缺少对老熟过程中窖泥微生物种群时序动态变化趋势的检测方法。种群变化定量方法的缺失,可能造成对种群作用解释的误判,如在肠道菌群组成研究中,研究人员发现当样品间的微生物生物量差异很大时,仅利用相对丰度和表型进行关联分析,将影响微生物菌群特征与定量数据(如生理参数或代谢物浓度)的相关性分析,产生结论误判[8-9]。尽管已有研究报道使用qPCR方法可对窖泥内的梭菌纲微生物进行定量[10],但窖泥内存在大量未培养微生物,基于qPCR的绝对定量方法需设计特异性引物,存在引物偏好性和物种局限性的问题,并不适用于对所有的窖泥微生物种群同时进行精确的定量。现阶段,利用已知浓度的DNA或细胞作为内标,基于高通量测序技术,发展出较为快捷的微生物绝对定量方法,在土壤菌群、肠道菌群、水体微生物、酿造微生物等研究领域已得到应用[9, 11-14]。本研究以不同老熟程度的窖泥内的窖泥菌群为研究对象,通过添加窖泥中不存在的丙酮丁醇梭菌(Clostridium acetobutylicum)作为内标菌,对窖泥微生物进行绝对丰度量化,基于窖泥微生物相对丰度与绝对丰度分析比较不同老熟程度下的窖泥菌群特征,为窖泥菌群结构认识到功能解析提供因果关系研究方法。
1 材料和方法 1.1 材料 1.1.1 窖泥:研究中所用的窖泥样品为不同窖龄的窖泥,取自安徽迎驾贡酒股份有限公司酿酒车间,取样位置为窖池底部(表 1),样品采集后放于含厌氧产气包的厌氧密封袋中运输保存。样品包括新窖泥(New)、趋老熟窖泥(Aging)和老熟窖泥(Aged)三种类型,每种类型样品各5个。样品分装后于–20 ℃保存,用于理化检测和窖泥DNA提取。
| Aging level of pit clay | Number of samples | Cellar age |
| New | 5 | 1–2 years |
| Aging | 5 | 4–8 years |
| Aged | 5 | Over 10 years |
1.1.2 主要试剂和仪器及培养基:
常规化学试剂购于国药集团化学试剂公司;标准品乙酸、丁酸、叔戊酸、己酸、乳酸购于美国Sigma-Aldrich试剂公司;厌氧密封袋与厌氧产气包(AnaeroPack)购于日本三菱瓦斯化学株式会社;PowerSoil DNA提取试剂盒购于美国Qiagen公司;细菌基因组DNA提取试剂盒购于中国捷瑞生物工程有限公司。
NanoDrop 8000蛋白核酸测定分光光度计、厌氧操作箱购于美国Thermo Fisher Scientific公司;气相色谱仪Agilent 7890B、高效液相色谱仪Agilent 1260购于美国Agilent公司;精密pH计购于美国Mettler Toledo公司;有机酸柱Aminex HPX-87H购于美国Bio-Rad公司。
CGM培养基(g/L):葡萄糖20,蛋白胨10,酵母粉10,(NH4)2SO4 2,K2HPO4 1,KH2PO4 0.5,MgSO4·7H2O 0.1,FeSO4·7H2O 0.015,MnSO4·H2O 0.01,CaCl2 0.01,ZnSO4 0.002,CoCl2 0.002,最终pH为7.0。
1.2 内标菌制备本研究选取窖泥中不存在的丙酮丁醇梭菌ATCC 824菌株作为内标参考菌,该菌株购买自美国典型培养物保藏中心(ATCC)。
按照5% (V/V)的接种比例接种丙酮丁醇梭菌过夜培养种子液至100 mL新鲜CGM培养基中,在厌氧操作箱中于37 ℃培养4–5 h至OD600 0.5–0.7,取菌液于12000 r/min离心10 min收集菌体,生理盐水重悬后用血球计数板计数,计数细胞浓度为1.07×109 cells/mL。一部分重悬菌体于–80 ℃冻存,作为内标细菌细胞待用;另一部分菌体参照细菌基因组DNA提取试剂盒说明书提取DNA,经NanoDrop 8000分光光度计测定DNA浓度后作为内标菌基因组DNA待用,将基因组DNA浓度稀释至工作浓度(本研究中基因组DNA的使用浓度为25.08 ng/μL)。
1.3 内标添加方式的评估方法添加内标的目的在于校正研究样本之间生物量的差异,以此标准评估内标添加方式的合理性。本研究中内标添加方式分为丙酮丁醇梭菌菌体细胞和基因组DNA两种内标添加方式。设置3个内标添加梯度Level-1、Level-2和Level-3,添加丙酮丁醇梭菌内标细胞的量比为1:10:50,每个浓度梯度设立2个平行,采用同一窖泥样本进行梯度内标添加验证,理论窖泥微生物绝对生物量比值为1:1:1。参照PowerSoil DNA试剂盒说明书,称取0.25 g窖泥于破碎管中并添加内标菌后进行基因组DNA提取。内标菌细胞取自1.2中制备的丙酮丁醇梭菌重悬菌液,最终3个梯度下添加的内标细胞量分别为1.42×106 cells/0.25g窖泥、1.42×107 cells/0.25g窖泥和7.10×107 cells/0.25g窖泥,对应的添加浓度分别为5.68×106 cells/g窖泥、5.68×107 cells/g窖泥和2.84×108 cells/g窖泥。
内标菌基因组DNA取自于1.2中已提取好的丙酮丁醇梭菌基因组DNA,其添加量由公式(1)换算所得。尽量保证两种添加方式中内标菌DNA量在提取时的加入量一致,最终3个内标梯度提取添加的内标菌基因组DNA浓度分别为6.27 ng/0.25 g窖泥、62.7 ng/0.25 g窖泥和313.5 ng/0.25 g窖泥,对应添加的基因组DNA浓度分别为25.08 ng/g窖泥、250.8 ng/g窖泥和1254 ng/g窖泥。
所需添加的内标基因组DNA浓度的换算按照公式(1)进行。
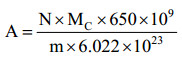
|
公式(1) |
式中,A为加入的DNA量(ng/g窖泥);m为用于提取基因组DNA的窖泥质量,本研究中窖泥质量m为0.25 g;N为添加的细菌细胞个数;MC为内标菌(丙酮丁醇梭菌)的基因组大小4.09 Mb;对于双链DNA,碱基对的分子质量为650 g/mol。
1.4 窖泥内有机酸分析本研究中所涉及的有机酸分析包括挥发性成分和非挥发性成分,挥发性成分主要为窖泥浸提液内的乙酸、丁酸和己酸,采用气相色谱法检测。非挥发性成分主要为窖泥浸提液内的乳酸,采用液相色谱法检测。
称取2 g左右的窖泥,添加8 mL超纯水,涡旋仪振荡10 s,超声30 min后12000 r/min离心10 min,保留上清液过0.22 μm水系滤膜后获得窖泥浸提液。取200 μL窖泥浸提液,加入50 μL浓度为12.5 g/L的叔戊酸内标溶液(含5%体积比的浓盐酸,pH 2.5),涡旋振荡30 s,之后10000 r/min离心5 min,吸取150 μL上清液进行气相色谱分析。色谱柱为Econo Cap-Wax毛细管柱(30 m×0.25 mm×0.25 μm),程序升温条件为:初始温度60 ℃,保持0.5 min,以20 ℃/min升到190 ℃,保持3 min。分流比为30:1,进样口温度220 ℃,氢火焰离子检测器(FID)检测温度220 ℃。
取200 μL窖泥浸提液,使用高效液相色谱仪分析系统测定乳酸,色谱柱为Aminex HPX-87H (Bio-Rad),流动相为5 mmol/L稀硫酸溶液,流速0.6 mL/min,柱温60 ℃,检测器为示差检测器[15]。
1.5 窖泥pH测定称量2 g左右窖泥,加入6 mL超纯水,涡旋振荡30 s后静置,12000 r/min离心10 min,取上清液用pH计直接测量pH[10]。
1.6 DNA提取与高通量测序窖泥基因组DNA参照PowerSoil DNA试剂盒说明书称取0.25 g窖泥于破碎管并添加内标后进行提取。PCR扩增引物为515modF (5′-GTGYC AGCMGCCGCGGTAA-3′)与806modR (5′-GGAC TACNVGGGTWTCTAAT-3′),对细菌和古菌的16S rRNA基因V4可变区进行扩增[16-17],建库与测序在上海美吉生物科技技术有限公司Illumina Miseq PE300测序平台完成。测序原始数据已上传NCBI,检索号(accession number)为PRJNA687810。
1.7 生物信息学分析原始序列经FLASH进行质控和过滤,去除低质量序列并将双端序列合并为单端序列,设置最小overlap长度为10 bp,拼接区允许最大错配率为0.2。提取样本barcode并切除引物区域,barcode允许的错配数为0,最大引物错配数为2,最终获得优化序列[18]。经Usearch提取非重复序列,按97%相似性对非重复序列进行可操作分类单元(operational taxonomic unit,OTU)聚类,去除嵌合体,获得OTU的代表序列。对应物种分类信息采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,比对数据库为Silva 132数据库[19-20]。
1.8 窖泥微生物的生物量计算方法经注释获得代表序列OTU统计表,根据OTU统计表内的内标相对丰度计算得到窖泥微生物的生物总量(公式2)。

|
公式(2) |
A1为窖泥微生物的生物总量(cells/g窖泥);N为每克窖泥所添加的内标菌细胞数(cells/g窖泥),n为内标菌(丙酮丁醇梭菌)的相对丰度(%)。
经计算获得窖泥微生物的生物总量后,接着对窖泥微生物菌群相对丰度组成百分化处理,经计算可得窖泥微生物某物种属的生物量(公式3)。

|
公式(3) |
A2为原位窖泥内某物种属的绝对丰度(cells/g窖泥);A1为通过公式(2)计算的原位窖泥微生物总量(cells/g窖泥);a为窖泥内某物种属的相对丰度(%)。
2 结果和分析 2.1 窖泥微生物定量方法的建立首先对内标的两种添加方式的定量适用性进行评估。扩增子测序结果表明(图 1),添加DNA为内标时,3个内标梯度Level-1、Level-2和Level-3的内标菌平均相对丰度分别为0.17%、0.77%、2.20%,对应比值为1:4.5:13。经公式(2)计算以DNA为内标时窖泥微生物的绝对生物量,3个内标梯度组的窖泥微生物绝对生物量比值为1:2.1:3.7,与理论值1:1:1相差甚远,对应的梯度组内窖泥微生物绝对生物量相对标准偏差(即变异系数)分别为22.97%、6.96%和16.68% (表 2)。

|
| 图 1 添加丙酮丁醇梭菌基因组DNA (A)和添加丙酮丁醇梭菌细胞(B)作为内标的窖泥微生物群落结构组成(属水平) Figure 1 Prokaryotic community structures with two types of internal controls. A: genomic DNA of Clostridium acetobutylicum was used as internal control; B: cells of Clostridium acetobutylicum were used as internal control. Level-1, Level-2 and Level-3 represent three internal gradients, and R1 and R2 were duplicates. |
| Sample | DNA amount/(ng/g) | Cells amount/(×106 cells/g) | Relative abundance of internal standard/(Mean±SD, %) | Absolute biomass in pit clay sample/(Mean±SD, ×108 cells/g) | Relative standard deviation of absolute biomass in each level/% |
| Adding genomic DNA | |||||
| Level-1 | 25.08 | – | 0.17±0.04 | 35.30±8.11 | 22.97 |
| Level-2 | 250.80 | – | 0.77±0.05 | 74.05±5.16 | 6.96 |
| Level-3 | 1254.00 | – | 2.20±0.36 | 129.74±21.64 | 16.68 |
| Adding cells | |||||
| Level-1 | – | 5.68 | 0.33±0.03 | 17.09±1.67 | 9.80 |
| Level-2 | – | 56.80 | 2.93±0.36 | 19.11±2.39 | 12.51 |
| Level-3 | – | 284.00 | 14.23±1.54 | 17.35±2.19 | 12.61 |
添加细胞作为内标时,3个内标梯度Level-1、Level-2和Level-3的内标菌平均相对丰度分别为0.33%、2.93%和14.23%,对应比值为1:8.9:43。经公式(2)计算以细胞作为内标时窖泥微生物的绝对生物量,3个内标梯度组的窖泥微生物绝对生物量比值为1:1.12:1.02,接近理论值1:1:1,对应的梯度组内窖泥微生物绝对生物量相对标准偏差(即变异系数)分别为9.80%、12.51%和12.61% (表 2)。以细菌细胞为内标时,总样本(3个梯度下6个样本)的窖泥微生物绝对生物量相对标准偏差为12.82%,远小于基因组DNA作为内标添加时51.57%的相对标准偏差值。
当采用同一窖泥样本验证内标的有效性时,添加细菌细胞作为内标时回收所得的内标的相对丰度比值更接近于理论设定的内标梯度比值(1:10:50),且获得的窖泥微生物绝对生物量接近理论比值1:1:1。可见,添加细菌细胞作为内标的方式的重复性与稳定性优于以基因组DNA作为内标的添加方式。同时,重复性实验表明,除内标菌外,同一个窖泥样本的菌群结构在不同提取组间保持基本一致,表明不同提取组和建库对菌群结构影响较为有限(图 1)。上述结果表明添加细菌细胞作为内标较添加DNA作为内标的绝对定量方法更适宜于窖泥微生物菌群结构定量分析。
2.2 不同老熟程度窖泥的菌群多样性采用高通量测序技术,对安徽迎驾贡酒股份有限公司酿酒车间内不同老熟程度的窖泥微生物菌群结构进行解析,添加丙酮丁醇梭菌细胞为内标。新窖泥(new)、趋老熟窖泥(aging)与老熟窖泥(aged)菌群的α多样性存在显著性差异。具体而言,随着窖泥的老熟,窖泥菌群的Shannon指数增大,新窖泥、趋老熟窖泥与老熟窖泥内菌群平均Shannon指数分别为0.29、0.69与2.34,老熟窖泥的Shannon指数分别为新窖泥与趋老熟窖泥的8倍与3.4倍,以老熟窖泥内的群落多样性最高(图 2-A)。基于unweighted-unifrac距离算法,对窖泥菌群结构进行主坐标分析(PCoA,principal co-ordinates analysis),比较不同老熟度窖泥微生物菌群的β-多样性。PCoA分析结果表明,主成分1和成分2解释了总群落52.09%的变量,ANOSIM组间差异检验结果R值为0.6542 (P=0.001),不同老熟程度窖泥内菌群结构β多样性差异显著。这些结果表明依据老熟程度对窖泥进行分组的合理性,且新窖泥变为老熟窖泥的过程中,趋老熟窖泥属于介于新窖泥与老窖泥之间的过渡态。同时,新窖泥与趋老熟窖泥组内样本的菌群组成差异大于老熟窖泥,表明即使处于同一老熟水平的不同窖池,其窖泥老熟速度仍具有差异,而当窖泥完全老熟后,窖泥菌群组成会变得更为相近(图 2-B)。

|
| 图 2 不同老熟度窖泥菌群多样性比较 Figure 2 The diversity of prokaryotic communities of pit clay at different aging status. A: Shannon index of α-diversity. B: Principal co-ordinates analysis of β-diversity. Sample 1 to 5 were new pit clay, sample 6 to 10 were aging pit clay, and sample 11 to 15 were aged pit clay (**: 0.001 < P≤0.01; ***: P≤0.001; determined by t-test). |
2.3 不同老熟程度窖泥菌群结构动态变化
如图 3所示,基于Silva (http://www.arb-silva.de/)分类学数据库注释,窖泥内的微生物主要分布为:细菌为厚壁菌门(Firmicutes)下的芽孢杆菌纲(Bacilli)和梭菌纲(Clostridia),以及拟杆菌门(Bacteroidetes)下拟杆菌纲(Bacteroidia);古菌为广古菌门(Euryarchaeota)下的甲烷杆菌纲(Methanobacteria)、甲烷微菌纲(Methanomicrobia)和热原体纲(Thermoplasmata)。窖泥老熟过程中相对丰度大于1%的属有61个,不同老熟程度窖泥内的优势属有所不同,新窖泥内以乳杆菌属(Lactobacillus)为主导,相对丰度最高可达77%。趋老熟的窖泥内则以乳杆菌属与己酸菌属(Caproiciproducens)共同作为优势菌属。老熟窖泥内则以己酸菌属、甲烷杆菌属(Methanobacterium)、no_rank_f_Clostridiaceae_1与甲烷八叠球菌属(Methanosarcina)为主要优势微生物。己酸菌属在老窖泥内占据绝对优势,相对丰度最高可达65%。窖泥菌群相对丰度的组成结果表明,窖泥在老熟过程中,菌群变化表现为:以乳杆菌属为单一优势属逐渐过渡到以己酸菌属为主,其他老熟窖泥指示菌如甲烷杆菌属、甲烷八叠球菌属、甲烷短杆菌属、互营单胞菌属[6-21]的相对丰度逐渐增加(图 3-A)。如图 3-B所示,根据换算公式(2)的绝对定量结果表明,老熟窖泥微生物平均生物量约为新窖泥和趋老熟窖泥的600倍和28倍,且趋老熟窖泥的生物量是新窖泥的20倍。可见老熟窖泥内的生物量显著高于其余两类窖泥,随着窖泥的老熟,窖泥微生物的生物量逐渐升高。

|
| 图 3 相对定量(A)与绝对定量(B)分析窖泥老熟过程的菌群结构 Figure 3 Relative abundance (A) and absolute abundance (B) of the dominant genera in pit clay. Sample 1 to 5 were new pit clay, sample 6 to 10 were aging pit clay and sample 11 to 15 were aged pit clay. |
为进一步更为直观地分析不同老熟程度窖泥内重要功能微生物乳杆菌属(产乳酸)、己酸菌属(产己酸)、甲烷八叠球菌属(产甲烷)、甲烷杆菌属(产甲烷)、甲烷短杆菌属(产甲烷)、互营单胞菌属(短中链脂肪酸降解)的差异,结合换算公式(3),以相对定量和绝对定量分析这些微生物在窖泥老熟中的变化趋势。
如图 4所示,随着窖泥的老熟,乳杆菌属的相对丰度在老熟过程中不断降低,但其绝对丰度却呈现完全相反的变化,即其绝对丰度在老熟过程中不断升高。老熟窖泥内的乳杆菌属平均绝对丰度可达2.71×107 cells/g窖泥,是新窖泥的20倍,为趋老熟窖泥的1.5倍(图 4-A)。己酸菌属是窖泥内主要的产己酸菌,在老熟窖泥中的平均绝对丰度可达5.90×108 cells/g,绝对丰度分别为新窖泥和趋老熟窖泥的25000倍和20倍。趋老熟窖泥样本中己酸菌属平均丰度为2.54×107 cells/g窖泥,高于新窖泥内的2.35×104 cells/g窖泥,约为后者的1000倍(图 4-B)。己酸菌属的相对丰度和绝对丰度均随着窖泥的老熟逐步升高,其生物量呈现出数量级增长的变化,最终在老熟窖泥内占据绝对优势。不同老熟程度窖泥内的互营单胞菌属相对丰度无显著差异,但对比绝对丰度,老熟窖泥内的绝对丰度可达1.30×107 cells/g窖泥,分别是新窖泥内与趋老熟窖泥的1250倍和150倍(图 4-C)。以甲烷杆菌属、甲烷八叠球菌属、甲烷短杆菌属为代表的甲烷菌是窖泥内主要甲烷菌,它们的相对丰度占比和绝对丰度随着窖泥的老熟不断升高:甲烷杆菌属在老熟窖泥内的平均绝对丰度可达2.15×108 cells/g窖泥,分别为新窖泥和老熟窖泥的48829倍和1790倍;甲烷八叠球菌属的平均绝对丰度为8.89×107 cells/g窖泥,分别为新窖泥和趋老熟窖泥的7362倍和2403倍;甲烷短杆菌属的平均绝对丰度为1.94×107 cells/g窖泥,分别为新窖泥和趋老熟窖泥的1077倍和35倍(图 4-D–F)。

|
| 图 4 主要功能窖泥微生物在不同老熟程度窖泥内的相对丰度和绝对丰度变化比较 Figure 4 The comparation of functional microbes in pit clay with different aging status by relative abundance analysis and absolute abundance analysis. A: Lactobacillus; B: Caproiciproducens; C: Syntrophomonas; D: Methanobacterium; E: Methanosarcina; F: Methanobrevibacter. The bar represents the mean value and the whisker represents the standard deviation. |
对重要功能菌的相对丰度与绝对丰度分析表明,相对丰度呈现的物种结构差异并不一定能真正反映实际的微生物绝对丰度变化趋势,如乳杆菌属、互营单胞菌属、甲烷八叠球菌属和甲烷短杆菌属。绝对丰度与相对丰度联用的方法可以为窖泥菌群结构和功能分析提供更为全面的信息。
2.4 不同老熟程度窖泥的理化表征与窖泥微生物相对及绝对丰度相关性进一步分析新窖泥、趋老熟窖泥、老熟窖泥的理化表征与主要菌属的关系。新窖泥、趋老熟窖泥内含有大量的乳酸,平均乳酸浓度分别为98.42 g/kg与51.70 g/kg,而老熟窖泥平均仅含1.20 g/kg的乳酸(图 5-A)。Spearman相关性表明窖泥内的乳酸含量与乳杆菌属相对丰度呈正相关(图 6-A),与乳杆菌属的绝对丰度呈负相关(图 6-B),老熟窖泥中的乳杆菌属绝对丰度分别是新窖泥与趋老熟窖泥内的20倍与1.5倍(图 4-A),但老熟窖泥中的乳酸含量却仅为新窖泥和趋老熟窖泥的1.2%和2.3%,表明老熟窖泥中较低的乳酸含量并非老窖泥中乳杆菌属绝对丰度较低导致。

|
| 图 5 不同老熟阶段窖泥理化指标 Figure 5 Physical and chemical characteristics of pit clay. A: lactic acid; B: pH; C: butyrate; D: caproate. Significance analysis was determined by t-test. *: 0.01 < P < 0.05; **: 0.001 < P≤0.01. The box represents the 25%–75% data range, vertical line represents the means±standard deviations (n=5), and the horizontal line represents the median value. |

|
| 图 6 理化因子与菌群丰度的Spearman相关性 Figure 6 Spearman correlation analysis of the physicochemical characteristics and the abundance of important prokaryotes at genus level. A: Correlation between relative abundance of genus and physicochemical characteristics. B: Correlation between absolute abundance of genus and physicochemical characteristics. *: 0.01 < P < 0.05; **: 0.001 < P≤0.01; ***: P≤0.001, the correlation significance. |
窖泥pH随着窖泥的老熟逐渐升高,新窖泥和趋老熟窖泥与老熟窖泥之间的pH存在显著差异,pH与窖泥内的乳酸含量呈显著负相关,表明不同老熟阶段下窖泥pH的变化与乳酸含量紧密相关,原因可能在于新窖泥与趋老熟窖泥的菌群结构相对简单,以可发酵还原糖产生大量乳酸的乳杆菌属为优势属,而新窖泥与趋老熟窖泥内的总体生物量远低于老窖泥,以己酸菌属为代表的乳酸利用菌可能在消减窖泥乳酸含量过程中具有重要作用[22-23],结合老熟窖泥中的己酸菌属含量分别为新窖泥和趋老熟窖泥的25000倍与20倍,由于新窖泥和趋老熟窖泥内的己酸菌属绝对丰度低,导致乳酸无法被消减因而大量积累,故新窖泥和趋老熟窖泥的pH较低(图 5-B)。相对定量分析显示丁酸与己酸菌属正相关(图 6-A),绝对定量分析却表明丁酸含量与己酸菌属不相关(图 6-B),结合丁酸在窖泥老熟过程中的变化倍数远小于己酸的变化(图 5-C–D),己酸菌属微生物产生丁酸能力较弱,故绝对定量的相关性分析结果相较于相对定量分析结果而言更与实际情况相符。己酸是己酸菌属的主要产物,随着窖泥的老熟,己酸菌属在窖泥内的丰度增大,己酸浓度与己酸菌属的相对丰度及绝对丰度均呈现出显著正相关(图 6),这与老熟过程中的窖泥pH由酸性逐步变化为近中性、适宜于己酸的生成相一致(图 5-D)。绝对定量分析表明,窖泥中己酸浓度与重点分析的6个属显著正相关,而相对定量分析仅识别出己酸浓度与己酸菌属及甲烷杆菌属显著正相关。就相对丰度而言,乳杆菌属是新窖泥与趋老熟窖泥内的优势微生物,与己酸菌属、甲烷菌、互营单胞菌属之间呈负相关(图 6-A);而以绝对丰度而言,窖泥内微生物量随着窖泥的老熟而增加,乳杆菌属与其余5个属在相关性结果上则显示出正相关的趋势(图 6-B)。
窖泥微生物生成的有机酸与窖泥微生物丰度之间的相关性,在采用相对定量丰度与绝对定量丰度时,表现出不尽相同的相关性结果。原因在于相对丰度关注的是菌群组成的变化,当生物量存在较大差异时,相对丰度高的微生物的绝对丰度不一定高,反之亦然,所以利用相对丰度和表型进行关联分析,可能会产生结论误判[8-9]。随着窖泥的老熟,老熟窖泥微生物平均生物量约为新窖泥和趋老熟窖泥的600倍和28倍,存在较大的生物量差异,因此微生物代谢产物的累积更多与微生物绝对丰度有更大的关系,表明在相关性分析时,不仅需要关注相对丰度变化,更应关注窖泥微生物的绝对丰度变化。如乳杆菌的相对丰度与乳酸浓度呈现正相关(图 6-A),即先前的研究一般推测新窖泥中高丰度的乳杆菌属导致了较高浓度的乳酸,但本研究表明窖泥中乳酸较低的更重要原因在于以己酸菌为代表的乳酸利用型菌株可以将乳酸及时降解,在老窖泥中呈现出乳杆菌绝对丰度更高但累积的乳酸浓度却更低的现象(图 6-B)。因此,结合绝对丰度与相对丰度的相关性分析结果,可在窖泥微生物代谢功能分析方面得出更准确的结论。
3 讨论和结论随着窖泥的老熟,窖泥内微生物多样性增加,其中风味物质产生菌的增加对生产优质原酒具有重要意义[24]。之前的研究多采用相对定量分析各类型微生物在各窖泥样本中的相对丰度,本研究利用高通量测序技术,通过外加内标菌进行定量的方式,对测序样本的菌体生物量进行校正,建立起窖泥微生物含量的绝对定量方法。基于相对丰度和绝对丰度对不同老熟程度的窖泥进行菌群结构分析,解析窖泥老熟过程中的菌群结构与生物量的动态变化,有助于加深我们对于窖泥老熟过程中窖泥微生物菌群结构变化的认识。
菌群结构分析的最终目的为分析不同种群的生物学功能,绝对定量方法可在分析不同种群功能方面比相对定量方法提供更为全面的信息。如乳杆菌属是新窖与退化窖泥内的特征微生物[7, 21],之前基于相对定量的分析认为大量的乳杆菌属不利于优质白酒的生产,但本研究的绝对定量分析表明窖泥老熟过程乳杆菌属生物量不断增加,老熟窖泥内的乳杆菌属绝对丰度并未降低,反而大幅度增加,这与通过相对定量得出的结论截然相反。因此我们推测新窖泥中乳杆菌相对丰度较高只是因为其他代表老熟窖泥的特征菌如己酸菌属、互营单胞菌属和甲烷菌,未能很好生长出来。此外,窖泥退化的原因可能并非乳杆菌的繁殖异常,而是因为其他老熟窖泥特征菌的衰亡[21]。因此,新窖泥和退化窖泥中乳杆菌相对丰度较高是老熟窖泥特征菌未能完全生长或退化的结果,而非原因。进一步而言,绝对定量分析表明老熟窖泥内虽然乳杆菌绝对量远高于新窖泥和趋老熟窖泥,因此老熟窖泥可能具有更高的乳酸产生能力,但老熟窖泥中能利用乳酸的微生物如己酸菌属绝对丰度也较高,使得老熟窖泥中的乳酸可以转化为己酸,促进窖泥pH稳态的维持。此外,甲烷菌与互营单胞菌属等酸降解菌在窖泥老熟过程中绝对丰度不断增加,也促进了窖泥稳态环境的维持[6-7, 21, 25-26]。
绝对定量的研究方法促进了我们对窖泥菌群结构与功能的认识,但不可否认的是,绝对定量的方法仍存在提取效率误差问题,该问题也同样困扰着其他领域的研究者们[13, 27]。因此,在研究过程中尽可能选择合适的内标与添加方式显得尤为重要[28]。本研究中细胞内标法定量的重复性和线性均优于直接DNA内标法,可能原因在于:有报道称土壤对细胞和胞外DNA的吸附程度不同[29],窖泥的基质为黏土,对直接添加的DNA内标可能也存在类似选择性吸附问题,小体积内标DNA溶液加入后难以在窖泥中达到较好的分散度,造成提取率不够均匀。本研究表明,选择的丙酮丁醇梭菌菌体细胞作为内标是较为合适的,只需在提取窖泥DNA前加入一定量的内标菌菌体,操作较为简便。
基于扩增子测序的相对定量方式有助于研究窖泥微生物多样性,比较不同类型窖泥微生物菌群的结构差异。本研究建立的绝对定量方法仅需在窖泥微生物DNA提取阶段添加内标菌,通过后续高通量扩增子测序即可同时对窖泥菌群进行相对丰度与绝对丰度分析。基于本研究的绝对定量方法,可以用于窖泥微生物功能分析,推进相关性分析升级到因果分析。此外,基于绝对定量的菌群结构分析也可以用于判定新制备的人工窖泥的质量,以及监测生产中窖泥质量变化趋势。
| [1] | 沈怡方. 白酒生产技术全书. 北京: 中国轻工业出版社, 1998. |
| [2] |
Ren C, Du H, Xu Y. Advances in microbiome study of traditional Chinese fermented foods. Acta Microbiologica Sinica, 2017, 57(6): 885-898.
(in Chinese) 任聪, 杜海, 徐岩. 中国传统发酵食品微生物组研究进展. 微生物学报, 2017, 57(6): 885-898. |
| [3] |
Hou XG, Wang JY, Li XS, Hu BY, Li SL, Gao YY. Research progress in main functional microbes in Nong-flavor liquor pits. Liquor-Making Science & Technology, 2013(2): 96-101, 106.
(in Chinese) 侯小歌, 王俊英, 李学思, 胡炳义, 李绍亮, 高应运. 浓香型白酒窖池主要功能性微生物的研究进展. 酿酒科技, 2013(2): 96-101, 106. |
| [4] | Gao JJ, Liu GY, Li AJ, Liang CC, Ren C, Xu Y. Domination of pit mud microbes in the formation of diverse flavour compounds during Chinese strong aroma-type Baijiu fermentation. LWT, 2021, 137: 110442. DOI:10.1016/j.lwt.2020.110442 |
| [5] | Liu MK, Tang YM, Guo XJ, Zhao K, Tian XH, Liu Y, Yao WC, Deng B, Ren DQ, Zhang XP. Deep sequencing reveals high bacterial diversity and phylogenetic novelty in pit mud from Luzhou Laojiao cellars for Chinese strong-flavor Baijiu. Food Research International, 2017, 102: 68-76. DOI:10.1016/j.foodres.2017.09.075 |
| [6] |
Tao Y, Rui JP, Li JB, Xu ZC, Li DP, Tang QL, Wei Y, Li XZ. Microbial community compositions and diversity in pit mud of Chinese Luzhou-flavor liquor. CIESC Journal, 2014, 65(5): 1800-1807.
(in Chinese) 陶勇, 芮俊鹏, 李家宝, 徐占成, 李大平, 唐清兰, 魏勇, 李香真. 浓香型白酒窖泥中细菌和古菌的组成与多样性. 化工学报, 2014, 65(5): 1800-1807. DOI:10.3969/j.issn.0438-1157.2014.05.034 |
| [7] | Tao Y, Li JB, Rui JP, Xu ZC, Zhou Y, Hu XH, Wang X, Liu MH, Li DP, Li XZ. Prokaryotic communities in pit mud from different-aged cellars used for the production of Chinese strong-flavored liquor. Applied and Environmental Microbiology, 2014, 80(7): 2254-2260. DOI:10.1128/AEM.04070-13 |
| [8] | Stämmler F, Gläsner J, Hiergeist A, Holler E, Weber D, Oefner PJ, Gessner A, Spang R. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome, 2016, 4(1): 28. DOI:10.1186/s40168-016-0175-0 |
| [9] | Vandeputte D, Kathagen G, D'hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, de Commer L, Darzi Y, Vermeire S, Falony G, Raes J. Quantitative microbiome profiling links gut community variation to microbial load. Nature, 2017, 551(7681): 507-511. DOI:10.1038/nature24460 |
| [10] | 胡晓龙. 浓香型白酒窖泥中梭菌群落多样性与窖泥质量关联性研究. 江南大学博士学位论文, 2015. |
| [11] | Lin YJ, Gifford S, Ducklow H, Schofield O, Cassar N. Towards quantitative microbiome community profiling using internal standards. Applied and Environmental Microbiology, 2019, 85(5): e02634-e02618. |
| [12] | Tkacz A, Hortala M, Poole PS. Absolute quantitation of microbiota abundance in environmental samples. Microbiome, 2018, 6(1): 1-13. DOI:10.1186/s40168-017-0383-2 |
| [13] | Yang L, Lou J, Wang HZ, Wu LS, Xu JM. Use of an improved high-throughput absolute abundance quantification method to characterize soil bacterial community and dynamics. Science of the Total Environment, 2018, 633: 360-371. DOI:10.1016/j.scitotenv.2018.03.201 |
| [14] | Du RB, Wu Q, Xu Y. Chinese liquor fermentation: identification of key flavor-producing Lactobacillus spp. by quantitative profiling with indigenous internal standards. Applied and Environmental Microbiology, 2020, 86(12): e00456-20. |
| [15] |
Gong LC, Ren C, Gao JJ, Xu Y. Construction of p-cresol reduction recombinant lactic acid bacterium to decrease p-cresol in Chinese liquor fermentation. Microbiology China, 2020, 47(3): 749-758.
(in Chinese) 宫璐婵, 任聪, 高江婧, 徐岩. 重组4-甲基苯酚消减乳酸菌的构建及其在白酒酿造体系中的功能. 微生物学通报, 2020, 47(3): 749-758. |
| [16] | Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell, 2016, 167(6): 1469-1480. DOI:10.1016/j.cell.2016.11.018 |
| [17] | Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, Gilbert JA, Jansson JK, Caporaso JG, Fuhrman JA, Apprill A, Knight R. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems, 2016, 1(1): e00009-e00015. |
| [18] | Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 2011, 27(21): 2957-2963. DOI:10.1093/bioinformatics/btr507 |
| [19] | Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 2013, 10(10): 996-998. DOI:10.1038/nmeth.2604 |
| [20] | Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research, 2013, 41(Database issue): D590-D596. |
| [21] | Hu XL, Du H, Ren C, Xu Y. Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese liquor fermentation pit muds. Applied and Environmental Microbiology, 2016, 82(8): 2506-2515. DOI:10.1128/AEM.03409-15 |
| [22] | Wang H, Li XZ, Wang Y, Tao Y, Lu SW, Zhu XY, Li DP. Improvement of n-caproic acid production with Ruminococcaceae bacterium CPB6:selection of electron acceptors and carbon sources and optimization of the culture medium. Microbial Cell Factories, 2018, 17(1): 1-9. DOI:10.1186/s12934-017-0850-2 |
| [23] |
Ren C, Gu Y, Du H, Xu Y. Predicting dominant caproate-producing microbes by comparing the microbiotas between new- and aged-pit muds. Food and Fermentation Industries, 2018, 44(12): 8-14.
(in Chinese) 任聪, 辜杨, 杜海, 徐岩. 基于新老窖泥的微生物菌群结构判定浓香型白酒生产中的主体己酸菌. 食品与发酵工业, 2018, 44(12): 8-14. |
| [24] | Wang CD, Chen Q, Wang Q, Li CH, Leng YY, Li SG, Zhou XW, Han WJ, Li JG, Zhang XH, Li YZ. Long-term batch brewing accumulates adaptive microbes, which comprehensively produce more flavorful Chinese liquors. Food Research International, 2014, 62: 894-901. DOI:10.1016/j.foodres.2014.05.017 |
| [25] | Guo MY, Huo DQ, Ghai R, Rodriguez-Valera F, Shen CH, Zhang N, Zhang SY, Hou CJ. Metagenomics of ancient fermentation pits used for the production of Chinese strong-aroma liquor. Genome Announcements, 2014, 2(5): e01045-e01014. |
| [26] | McInerney MJ, Bryant MP, Pfennig N. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Archives of Microbiology, 1979, 122(2): 129-135. DOI:10.1007/BF00411351 |
| [27] | Wu ZB, Wang SY, Zhang QQ, Hao JW, Lin YY, Zhang JY, Li AH. Assessing the intestinal bacterial community of farmed Nile tilapia (Oreochromis niloticus) by high-throughput absolute abundance quantification. Aquaculture, 2020, 529: 735688. |
| [28] | Harrison JG, John Calder W, Shuman B, Alex Buerkle C. The quest for absolute abundance: The use of internal standards for DNA-based community ecology. Molecular Ecology Resources, 2021, 21(1): 30-43. |
| [29] | Frostegård A, Courtois S, Ramisse V, Clerc S, Bernillon D, Le Gall F, Jeannin P, Nesme X, Simonet P. Quantification of bias related to the extraction of DNA directly from soils. Applied and Environmental Microbiology, 1999, 65(12): 5409-5420. |