 2022, Vol. 62
2022, Vol. 62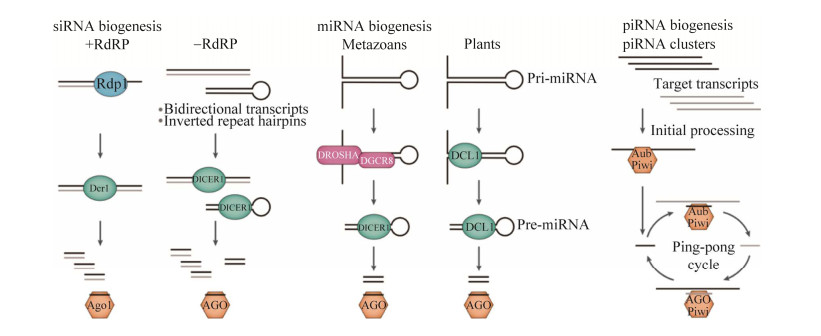





高通量分析技术在资源微生物及功能基因发掘中的应用

http://dx.doi.org/10.13343/j.cnki.wsxb.20220299
中国科学院微生物研究所,中国微生物学会
中国科学院微生物研究所,中国微生物学会
文章信息
- 张焕, 姜卫红, 顾阳. 2022
- ZHANG Huan, JIANG Weihong, GU Yang.
- 高通量分析技术在资源微生物及功能基因发掘中的应用
- Application of high-throughput techniques in the discovery of microbial resources and functional genes
- 微生物学报, 62(11): 4234-4246
- Acta Microbiologica Sinica, 62(11): 4234-4246
-
文章历史
- 收稿日期:2022-04-22
- 修回日期:2022-06-30
- 网络出版日期:2022-07-25
引用本文 |
张焕, 姜卫红, 顾阳. 高通量分析技术在资源微生物及功能基因发掘中的应用[J]. 微生物学报, 2022, 62(11): 4234-4246.
Zhang Huan, Jiang Weihong, Gu Yang. Application of high-throughput techniques in the discovery of microbial resources and functional genes[J]. Acta Microbiologica Sinica, 2022, 62(11): 4234-4246.
高通量分析技术在资源微生物及功能基因发掘中的应用
1. 中国科学院分子植物科学卓越创新中心, 植物生理生态研究所, 合成生物学重点实验室, 上海 200032;
2. 中国科学院大学, 北京 100049
2. 中国科学院大学, 北京 100049
摘要:随着新的微生物资源不断被发现以及微生物基因组测序数据的积累和完善,目前研究重点和难点是如何从大量数据中快速发现和鉴定与微生物重要表型相关的功能基因,这就需要高通量的分析研究手段,主要涉及建库和筛选两个主要技术单元。其中,建库是指构建能够覆盖微生物全基因组的突变或干扰文库,所涉及的技术包括宏基因组、转座子插入突变、RNA干扰(RNA interference,RNAi)、反转录子文库重组工程(retron library recombineering,RLR)、CRISPR抑制(CRISPRi)和CRISPR激活(CRISPRa)等。筛选则是通过某种胁迫压力来促使文库菌群的差异化生长,并结合高通量测序全面发掘与特定表型相关的功能基因,从而为后续研究提供有效信息。本文对功能基因组学研究中现有的高通量分析技术进行了梳理、总结和展望,以期为这类技术方法的拓展、优化以及应用提供参考。
关键词:微生物 功能基因组 高通量分析技术 文库构建 筛选
Application of high-throughput techniques in the discovery of microbial resources and functional genes
1. Key Laboratory of Synthetic Biology, Center for Excellence in Molecular Plant Sciences, Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai 200032, China;
2. University of Chinese Academy of Sciences, Beijing 100049, China
2. University of Chinese Academy of Sciences, Beijing 100049, China
Abstract: In light of the continuous discovery of microbial resources and the accumulating microbial genome data, it is an urgent task to screen and identify functional genes associated with important microbial phenotypes from the enormous data, for which high-throughput techniques are indispensible. The techniques are mainly involved in library construction and screening. Library construction refers to the establishment of mutant or interference libraries covering the whole genome of target microorganisms with techniques such as metagenomics, transposon insertion, RNA interference (RNAi), retron library recombineering (RLR), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa). As for the screening, a certain stress is often used to allow the differential growth of microbial individuals of a mutant library and then the causal relationships between specific genes and phenotypic outcomes at genomic level are identified by high-throughput sequencing. In this review, we briefly summarize the current high-throughput techniques used in functional genomics research, hoping to provide a reference to the development and optimization of these techniques in the future.
Keywords:
microorganisms functional genomics high-throughput techniques library construction screening
随着人类基因组计划的完成,生命科学的研究进入后基因组时代,进而产生了功能基因组学[1]。功能基因组学是利用基因组序列信息,通过相关的实验技术,在全基因组水平上研究基因的功能,并进行基因注释,建立基因型和表型之间的关系。功能基因组学的研究将生命科学的关注点由之前的单一基因转变为多基因,有助于阐明功能基因之间的相互关系及形成的网络,加速了对基因转录、翻译和调控等生命过程的认识,也有利于发掘新的生物资源。
微生物包括细菌、病毒、真菌以及一些小型的原生生物等,它们个体微小,但与人类关系密切。近年来,微生物功能基因组学的研究取得了很大进展。研究者们鉴定了大量的微生物功能基因,并利用合成生物学技术设计和改造细胞,这些研究不仅聚焦于工业微生物的性能优化,也涉及病原微生物的耐药性和致病性等方面。然而,相对于已经获得的海量微生物基因组数据,目前在功能基因发掘和利用方面还很不够,亟待加速跟进。例如,最常用的模式微生物大肠杆菌中仍有20%–30%的蛋白质编码基因功能未知[2–3];而其他微生物中未解析功能的基因数目则更多。显然,全面、高效地发掘和鉴定未知的重要功能基因,需要高通量的研究手段。目前,研究者们已经开发了一系列适用的高通量实验技术,从而为功能基因组学研究奠定了基础。
1 基于宏基因组学的微生物资源发掘“宏基因组学”(metagenomics)又被称为微生物环境基因组学或元基因组学。其通过提取特定环境样本中全部微生物的DNA构建宏基因组文库并结合下一代测序(next generation sequencing,NGS)技术,实现对样品所包含全部微生物的遗传组成及群落功能的分析,可用于高效发掘新的微生物菌种资源或功能基因。众所周知,自然界中微生物种类繁多,但实验室可培养的微生物物种有限。因此,利用宏基因组学的研究策略可快速挖掘海洋、土壤、肠道和冻土等环境中的不可培养微生物以及基因资源。目前,宏基因组学研究已被广泛地应用于医药、农业、遗传学和生物技术等多个领域[4]。
宏基因组学技术的一个重要应用场景是深海微生物资源的开发。由于深海高压低氧的环境特点,很多微生物无法在实验室条件下培养,因此,需要借助宏基因组文库来发现新的微生物物种和发掘新的生物元件。例如,2008年,研究人员通过构建宏基因组文库发现了一种生活在深海中的新型蓝藻,它由于缺乏光系统Ⅱ不能固碳,但却可以有效固氮,由此可能影响海洋碳氮平衡[5]。此外,研究人员从韩国西海岸的泥浆中提取细菌的总DNA,通过构建宏基因组文库筛选到一种新型的锌依赖的蛋白酶,该酶可有效水解偶氮酪蛋白和纤维蛋白,因此具有治疗血栓的潜力[6]。研究人员还利用宏基因组文库从海洋泥浆的微生物中筛选和鉴定了可用于生产虾青素的新型酯酶Est3-14,该酯酶的发现解决了使用皂化法生产虾青素产生许多副产品的问题[7]。
土壤生境下微生物的种类繁多,因此土壤中也存在大量的微生物资源有待开发。2017年,Ausec等收集酸性沼泽土壤环境中的微生物,通过对构建的宏基因组文库筛选,获得一种酸杆菌漆酶样多铜氧化酶(laccase-like multicopper oxidase,LMCO),该酶可以耐受高盐和高热的极端条件,因此具有很好的工业应用价值[8]。2021年,Yan等也通过类似技术从土壤微生物中筛选到一种新的微嗜酸、中度嗜热和对短链脂肪酸具有高活性的羟酸酯酶EstQ7,该酶作为生物催化剂,可应用于食品等多个工业领域[9]。
2 基于转座子突变的功能基因高通量筛选转座子是一段位于染色体上可自主复制并且可移动的DNA序列,自20世纪40年代美国科学家芭芭拉·麦克林托克首次在玉米中发现这种跳跃基因以来[10],科研人员开展了大量的研究工作,逐步揭开了转座子的神秘面纱。转座子按转座方式可大致分为两类,即Ⅰ型转座子(class Ⅰ elements)和Ⅱ型转座子(class Ⅱ elements)[11]。Ⅰ型转座子通过“复制-粘贴”的方式整合入基因组;Ⅱ型转座子主要通过“剪切-粘贴”的方式进行转座;少部分通过滚环(rolling circle)的方式转座,如helitron转座子;或通过其他未知机制转座,如polinton转座子和mavericks转座子[12]。目前研究人员在细菌、真菌以及高等生物中均发现了转座子的存在。
转座子突变的一个重要应用场景是建立突变体文库,并结合高通量实验技术全面发掘特定表型相关的功能基因。例如,基于转座子可在基因组随机插入的特性,早期研究人员利用融合了碱性磷酸酶(Tn-alkaline phosphatase,TnphoA)的转座子构建了霍乱弧菌的突变体文库,通过碱性磷酸酶作为报告基因分离出600多株突变菌株,并利用其进一步发掘与霍乱毒素表达相关的基因[13]。也有研究者在肺炎链球菌中构建了转座子随机突变体文库,并结合微阵列杂交技术在全基因组范围内快速鉴定功能基因[14]。显然,这些技术的应用可有效提高发现病原菌中致病性相关基因的效率。2009年,基于转座子突变的Tn-seq技术首次被报道(图 1)。此后,这一技术经过不断完善,在功能基因研究、代谢网络解析、基因互作和微生物复杂抗性等领域获得广泛应用[15]。

|
| 图 1 Tn-seq示意图[15] Figure 1 Schematic depiction of Tn-seq[15]. A: a gene disruption library is constructed by first transposing the mini-transposon magellan6, which contains an Mme I restriction site within each inverted repeat, into bacterial genomic DNA in vitro and then transforming a bacterial population with the transposed DNA. B: DNA is isolated from a portion of the bacterial pool (t1) and another portion is used to seed a culture on which selection is performed, then DNA is isolated again from recovered bacteria (t2). C: DNA from both time points is digested with Mme I. D: a PCR amplification was performed to obtain a 160 bp sequence with 20 bp of bacterial-specific DNA flanked by Illumina-specific sequences, which enable sequencing. After sequencing, different samples are identified based on barcode sequence, and the 20 bp reads are mapped to the genome and are counted for each insertion, thus allowing fitness to be calculated. |
Tn-seq技术最主要的一个应用是发掘必需基因。必需基因是维持生物体生长发育的关键基因,一旦基因发生扰动,其正常生长便会受到影响。因此,在突变体文库构建过程中需要确保较高的覆盖率,而在后续对该突变体库的高通量测序结果中,未检测到插入突变的基因序列即可被视为该生物体的必需基因。例如,2021年研究者在鲍曼不动杆菌中,分别构建了Himar1 mariner和Tn10-ATS (Tn10-altered target specificity)2个转座子文库,并在营养丰富培养基的条件下进行筛选,结果表明有372个基因在2个文库中均没有转座子插入,因而将这些基因定义为该菌生长的必需基因[16]。随后,研究者在脓肿分枝杆菌[17]、鳗弧菌[18]和短双歧杆菌[19]中相继构建了转座子突变体文库,用于鉴定菌株的必需基因。此外,Tn-seq方法也可用于条件必需基因的筛选。条件必需基因是指生物体在特定培养条件下维持生长发育的关键基因。研究者利用特定培养条件(如特定碳源、抑制物等)实现突变体文库菌株的竞争性生长,进而对菌群基因组进行高通量测序,通过与对照条件下菌群基因组测序结果的比对,即可全面地发掘该培养条件下的必需基因。例如,利用转座子文库,研究者在结核分枝杆菌中筛选了胆固醇代谢所需的必需基因[20],在霍乱弧菌中筛选到抵御内酰胺类抗生素对细胞壁破坏的双组分系统[21],以及在铜绿假单胞杆菌中鉴定与耐旱相关的基因等[22]。
尽管基于转座子的随机突变体文库可用于高通量的功能基因组学研究,但该方法也存在一些局限性和不足。一个主要的问题是转座子通常在基因组上存在插入热点,会导致插入位点在基因组上分布不均匀,因此一些短序列基因或小RNA编码序列无法被插入突变,进而在后续筛查过程中被遗漏。此外,转座子突变是随机性的,很难设计靶向特定的基因集群。
3 基于RNAi的功能基因高通量筛选RNAi是双链RNA (double-stranded RNA,dsRNA)诱导的转录后基因沉默现象。当dsRNA进入细胞后,被核糖核酸酶识别并剪切为21–23 bp的小干扰RNA (small interfering RNA,siRNA)。siRNA在核酸酶复合物的协助下,可与靶基因转录的信使RNA互补配对并导致其降解,从而实现对靶基因表达的干扰[23]。根据dsRNA的前体、合成途径以及装载蛋白的种类,可以将小RNA分为siRNA、miRNA (microRNA)以及piRNA (piwi-interacting RNA)[24] (图 2)。
随着对RNAi技术的深入了解,研究者建立了RNAi文库用于高通量的功能基因组学研究。2022年,Liu等使用Dharmacon siGenome文库,在胶质母细胞瘤干细胞全基因组范围内进行RNAi筛选,鉴定到ZNF117是肿瘤细胞发展成少突胶质细胞样肿瘤的主要调节因子,当抑制ZNF117的表达时,癌细胞会发展成更成熟的形式且易受到化疗的影响,为胶质母细胞瘤的治疗提供新的潜在靶点[25]。RNAi文库还可应用于细胞的耐药性筛选。例如,曲妥珠单抗可用于乳腺癌和胃癌患者的治疗,但耐药性的问题极大影响治疗效果。2013年,Boyer等使用定量蛋白质组学的方法分析抗曲妥珠单抗乳腺癌细胞的蛋白质变化,并根据差异表达的蛋白定制了小型的siRNA文库用于筛选与耐药性相关的蛋白[26]。在植物细胞中,研究人员通过农杆菌介导,将RNAi文库转化水稻的愈伤组织,用于筛选导致水稻分蘖数目减少和生长发育有关的基因[27]。此外,RNAi文库也被用于筛查模式植物拟南芥中抵御植物维管病原体的功能基因[28]。
对于微生物而言,RNAi技术被广泛应用于真菌和细菌等领域。在真菌的研究中,1994年,Cogoni等将类胡萝卜素合成所需基因转入到粗糙脉孢菌中,发现30%的转化菌株出现自身基因失活的现象,并将这一现象称之为基因抑制(gene quelling)[29]。随后,研究者在研究真菌中基因抑制现象的分子机制时,发现其与动物中的基因干扰机制极为类似,因此也将其归为RNAi[30]。2002年,Liu等首次建立了基于RNAi的真菌基因编辑技术,并用该技术成功失活了新型隐球菌的功能基因[31];随后,该技术陆续在烟曲霉菌和大丽轮枝菌等多种真菌中得以应用。
近年来,随着研究的深入,人们发现细菌中的sRNA(small RNA)作为基因表达的调控因子,不仅可以发挥基因抑制的作用,还可参与转录激活、mRNA的翻译和稳定性调节等过程[32]。2011年,Man等通过分析大肠杆菌中天然的sRNA结构特征,在细菌中首次设计人工反式编码sRNAs (artificial trans-encoded sRNAs,atsRNAs)元件,并利用该元件有效地抑制大肠杆菌中外源egfp基因和内源uidA基因的表达。但遗憾的是,该系统在革兰氏阳性菌如金黄色葡萄球菌中不能有效发挥抑制作用[33]。RNAi技术除可用于研究细菌的功能基因外,还可应用于代谢工程的改造中。2013年,Na等在大肠杆菌中使用RNAi技术抑制酪氨酸旁路代谢途径中tyrA和csrA基因的表达,使得菌株酪氨酸产量得到提升;作者进一步构建了122个靶向尸胺合成和调控途径的RNAi文库,利用该文库筛选,发现抑制ackA和pdhR基因的表达可使尸胺的产量分别提高40.2%和31.4%[34]。然而,目前细菌中RNAi的脱靶率仍然较高[33],因此并不适合构建大规模的干扰文库用于高通量筛选,亟待后续的优化改造。
综上所述,RNAi文库在高通量功能基因组学研究中具有独特优势,包括操作简单,成本较低,且可在文库设计时靶向特定的基因集群。但同时,该方法也存在着一定的不足,主要是脱靶率较高,且由于是在mRNA水平上抑制基因的表达,无法实现基因的完全失活,会导致筛选结果中出现较多的假阴性。
4 基于CRISPR-Cas系统的功能基因高通量筛选1987年,Ishino等在大肠杆菌中发现高度同源序列重复性出现,且这些重复序列被32个碱基间隔开[35],当时并不知道这些序列的功能。直到2002年,Jansen等发现该特征序列仅存在于细菌和古菌中,并将其命名为规律间隔成簇短回文重复序列(clustered regularly interspaced short palindromic repeats),即CRISPR,而临近CRISPR的基因则命名为Cas (CRISPR-associated)[36]。2008年,科学家证实了CRISPR-Cas系统是一种抵御外源DNA入侵的适应性免疫系统[37],其发挥作用包括3个阶段,第一阶段为外源DNA的捕获;第二阶段为CRISPR RNA (crRNA)的合成;第三阶段为目的基因的靶向[38]。2012年,研究人员证明该系统具有体外特异性切割双链DNA的能力[39],这一发现促进了CRISPR-Cas系统被开发为强大的基因组编辑工具(图 3)。
CRISPR-Cas系统的一个重要应用是建立大容量的基因CRISPR抑制(CRISPRi)或CRISPR激活(CRISPRa)文库,用于高通量的功能基因筛查,这两种文库都需借助失去核酸内切酶活性的Cas蛋白(dCas)发挥功能。其中,CRISPRi是利用向导RNA将dCas蛋白靶向目标基因的启动子区域以阻止RNA聚合酶结合和起始转录,或靶向基因编码框以阻止转录延伸;而CRISPRa则是利用向导RNA将融合dCas以及转录激活因子的蛋白复合体靶向目标基因的启动子上游区域,进而促进基因的转录[40]。
在哺乳动物细胞中,CRISPR文库可用于高通量筛选癌症治疗相关的功能基因。2021年,Wang等建立了小鼠慢病毒CRISPR-Cas9敲除(MusCK)文库,将该文库转导到4T1 (小鼠乳腺癌)细胞中,靶向肿瘤发生、发展和免疫调节相关的4 500个基因,并将这些细胞植入不同背景的小鼠中,筛选到癌细胞E3泛素连接酶Cop1可作为巨噬细胞浸润和免疫治疗的靶点[41]。此外,研究者还利用CRISPR-Cas系统开发了基因抑制或激活文库。2019年,Bassaganyas等将dCas9融合表达Kox1的转录阻遏物结构域KRAB蛋白,在HeLa细胞中构建覆盖全基因组的CRISPRi文库,筛选蛋白质转运相关的功能基因[42]。2014年,Gilbert等使用分子挂钩(SunTag)技术,将多拷贝的单纯性疱疹病毒蛋白16的合成四聚体(VP64) “挂到” dCas9蛋白上,构建覆盖全基因组的CRISPRa文库,鉴定与癌细胞生长以及调控组织发育相关的功能基因[43]。
对于微生物而言,目前的报道主要是基于CRISPRi文库筛选功能基因的研究,尚未有CRISPRa文库的相关研究报道。2018年,我国的科研人员建立了覆盖大肠杆菌全基因组的CRISPRi文库,并结合高通量测序技术,鉴定了一系列与糠醛(木质纤维素水解液中的一种主要抑制物)耐受相关的功能基因以及生长必需基因[44];Rousset等构建CRISPRi文库,在全基因组范围内筛选大肠杆菌中与噬菌体感染相关的宿主因子,为噬菌体-宿主之间的相互作用提供新的见解[45]。除了全基因组规模的大范围筛选,CRISPRi文库也适用于筛选特定功能基因集。2016年,Peters等在枯草芽孢杆菌中构建了针对289个必需基因的CRISPRi文库,探究必需基因的功能。同时利用化学基因组学建立必需基因之间的互作关系[46]。随后,也有研究人员在肺炎链球菌中构建了348个潜在必需基因的CRISPRi文库,发现一些新的必需基因[47]。必需基因对菌株的生长至关重要,而调控网络和转运子等对工程菌的改造也发挥着重要的作用。2020年,Liu等使用基于CRISPR的可追踪基因组工程(CRISPR-enabled trackable genome engineering,CREATE),在大肠杆菌中构建调控网络文库,筛选异丁醇耐受相关的基因,并通过改造功能基因获得异丁醇高产菌株[48]。2022年,Liu等在产脯氨酸的谷氨酸棒杆菌中构建靶向所有转运子的CRISPRi文库,首次鉴定了谷氨酸棒杆菌中的脯氨酸转运子,并通过在高产脯氨酸的谷氨酸棒杆菌中过表达该基因,进一步提高了脯氨酸的产量[49]。虽然CRISPRi文库为全基因组范围内筛选功能基因提供便利,但构建文库需要设计并合成大量的向导RNAs (guide RNAs,gRNAs),费用较高且需要耗费较多的人力和时间。因此,美国哥伦比亚大学的研究者利用化脓性链球菌的CRISPR-Cas免疫机制,建立了CRISPR适应性介导的文库加工(CRISPR adaptation-mediated library manufacturing,CALM)方法,将宿主细胞变成了可生产crRNA的工厂,进而形成覆盖基因组的CRISPRi文库,并利用该文库筛查和鉴定了影响氨基糖苷敏感性的代谢途径[50]。此外,在自养细菌方面,本课题组聚焦于可利用一碳气体(CO2和CO)的食气梭菌,前期开发了多套基于CRISPR-Cas的基因编辑工具,用于食气梭菌功能基因的敲除或抑制[51–52];并基于CRISPR-ddCas12a基因抑制方法,建立了针对食气梭菌全部转录因子的CRISPRi文库,用于高效筛选与特定表型相关的转录因子,从而为菌株的优化改造提供借鉴。
在现有的高通量微生物功能基因组学研究中,利用CRISPR-Cas系统构建各种文库用于功能基因筛选,具有明显的优势:(1) CRISPR-Cas基因组编辑技术已经在微生物中得到极大地推广和应用,这就为构建各类文库奠定了技术基础。(2) 可以用于构建基因失活、抑制或激活等多种类型的文库。(3) 构建靶向不同基因位点的质粒时仅需改变20 bp左右的向导RNA序列,文库设计比较简单。(4) 可根据需要设计靶向特定功能的基因集。因此,尽管这一技术也存在一些不足,如脱靶和渗漏等,但仍不失为目前高通量功能基因组学研究的首选方法。
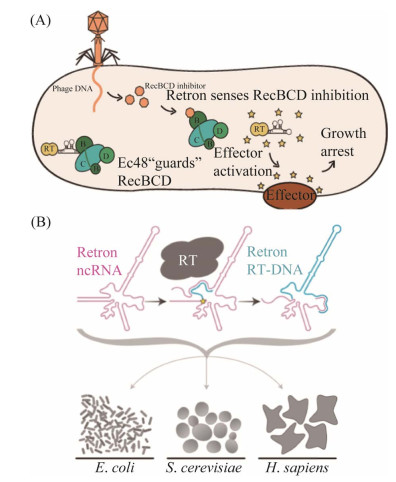
5 基于反转录子(retron)的功能基因高通量筛选1984年,Yee等首次在黄色粘球菌中发现了许多单链DNA短序列散布在细菌细胞中[53]。后续的研究发现,这些单链DNA片段都会与带有互补碱基序列的RNA相连,形成一个由RNA、DNA和酶组成的复合体,并证实该复合体是一种新的阻止噬菌体感染的防御系统,被命名为Retron[54]。2020年,以色列魏茨曼科学研究所的研究人员证实该系统由逆转录酶和非编码RNA组成,并首次解析了大肠杆菌中反转录子Ec48发挥作用的机制[55]。Ec48可用于检测RecBCD蛋白的完整性,当外源噬菌体感染大肠杆菌时,噬菌体蛋白会抑制RecBCD,进而诱导激活胞内Ec48的效应蛋白。该效应蛋白为跨膜蛋白,被激活后可改变细胞膜的通透性,从而导致大肠杆菌顿挫感染和细胞死亡,避免噬菌体大量增殖(图 4A)。2022年,Lopez等构建了大肠杆菌的Eco1 (大肠杆菌中的一种retron)突变体,发现retron突变体可促使模板DNA量增加8到10倍,并首次将该突变体应用于真核酵母细胞和人类细胞中进行基因编辑(图 4B)[56]。
随着对retron作用机制的深入了解,研究者进一步将其开发成适用于微生物的基因编辑工具。2021年,哈佛大学的Schubert等将retron用于创建大肠杆菌的基因突变体文库,并将这一方法命名为RLR。该方法将含有目标突变的反转录子序列与逆转录酶编码基因构建在同一质粒上,并导入大肠杆菌中表达,反转录子产生的携带目标突变的单链DNA可整合到子代细胞的基因组中,实现对基因组的编辑。为了进一步提高基因编辑效率来满足构建突变体文库的需求,作者阻断了大肠杆菌的天然错配修复机制,同时敲除外切核酸酶。在此基础上,构建了高效的RLR用于筛选大肠杆菌中与利福平耐药性相关的功能基因[57]。
RLR技术具有很多优势,包括高灵敏度和精确度、避免了Cas蛋白对宿主微生物潜在的毒性问题和极低的脱靶效应等。不过,目前该技术仅在大肠杆菌中被测试和使用,且可用的retron系统也较局限,有待未来进一步拓展和完善。
6 总结与展望得益于高通量实验技术的不断完善和发展,微生物功能基因组学的研究取得较大进展,并在微生物新资源的发掘中发挥着越来越重要的作用,极大促进了相关学科的发展。现有主要的几种文库构建方法各有特点,但也存在或多或少的不足。基于转座子随机突变的特点可用于构建全基因组的突变体文库,但不可避免地存在热点插入,造成覆盖度不佳的问题,且由于不同类型转座子对识别的DNA位点具有碱基偏好性,因此在采用该技术建库时需要考虑宿主菌基因组序列的碱基构成情况。RNAi技术既可在全基因组范围内也可在特定基因集范围内建立文库用于高通量筛选,但已有研究表明,RNAi技术具有较高的脱靶率,假阳性和假阴性结果较多,加大了后续实验验证的工作量。近年来发展起来的基于CRISPR-Cas系统的文库,一定程度地弥补了上述两种方法的不足,且成为了微生物功能基因组学研究的首选。但在微生物领域目前报道的主要是CRISPRi文库,而CRISPRa文库的研究进展较慢,这主要受制于基因转录激活元件的匮乏和激活效率的问题。最新报道的基于retron的突变体文库具有特定的优势,有望成为高通量功能基因组学研究中的一把新的利刃。
最后需要指出的是,目前微生物功能基因组学研究的限制瓶颈是可用的“筛子”极为有限,造成可筛选的表型屈指可数,主要局限于生长、耐受力以及少量具有可视特征(如颜色)的产品。尤其是在工业微生物领域,利用高通量筛选方法研究并提高目的产物的产量依旧是一个难题,即便有些产物具有颜色变化等特征,其显性变化范围也仅限于低浓度情况,并不能满足发掘高产菌的要求。因此,拓展和完善现有的筛选方法是微生物功能基因组学研究中需要重视的一个方向。
References
| [1] | Hagymási K, Tulassay Z. The Human Genome Project, genetic viability and genetic epidemiology. Orvosi Hetilap, 2005, 146(51): 2575-2580. |
| [2] | Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, Palsson BØ. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Molecular Systems Biology, 2011, 7: 535. DOI:10.1038/msb.2011.65 |
| [3] | Liang P, Labedan B, Riley M. Physiological genomics of Escherichia coli protein families. Physiological Genomics, 2002, 9(1): 15-26. DOI:10.1152/physiolgenomics.00086.2001 |
| [4] | Garrido-Cardenas JA, Manzano-Agugliaro F. The metagenomics worldwide research. Current Genetics, 2017, 63(5): 819-829. DOI:10.1007/s00294-017-0693-8 |
| [5] | Zehr JP, Bench SR, Carter BJ, Hewson I, Niazi F, Shi T, Tripp HJ, Affourtit JP. Globally distributed uncultivated oceanic N2-fixing cyanobacteria lack oxygenic photosystem Ⅱ. Science, 2008, 322(5904): 1110-1112. DOI:10.1126/science.1165340 |
| [6] | Lee DG, Jeon JH, Jang MK, Kim NY, Lee JH, Lee JH, Kim SJ, Kim GD, Lee SH. Screening and characterization of a novel fibrinolytic metalloprotease from a metagenomic library. Biotechnology Letters, 2007, 29(3): 465-472. DOI:10.1007/s10529-006-9263-8 |
| [7] | Lu P, Gao XW, Dong H, Liu Z, Secundo F, Xue CH, Mao XZ. Identification of a novel esterase from marine environmental genomic DNA libraries and its application in production of free all- trans-astaxanthin. Journal of Agricultural and Food Chemistry, 2018, 66(11): 2812-2821. DOI:10.1021/acs.jafc.7b06062 |
| [8] | Ausec L, Berini F, Casciello C, Cretoiu MS, Van Elsas JD, Marinelli F, Mandic-Mulec I. The first acidobacterial laccase-like multicopper oxidase revealed by metagenomics shows high salt and thermo-tolerance. Applied Microbiology and Biotechnology, 2017, 101(15): 6261-6276. DOI:10.1007/s00253-017-8345-y |
| [9] | Yan ZZ, Ding LP, Zou DD, Wang LY, Tan YZ, Guo ST, Zhang YC, Xin ZH. Identification and characterization of a novel carboxylesterase EstQ7 from a soil metagenomic library. Archives of Microbiology, 2021, 203(7): 4113-4125. DOI:10.1007/s00203-021-02398-0 |
| [10] | McClintock B. The significance of responses of the genome to challenge. Science, 1984, 226(4676): 792-801. DOI:10.1126/science.15739260 |
| [11] | Wang JW, Huang JJ, Shi G. Retrotransposons in pluripotent stem cells. Cell Regeneration: London, England, 2020, 9(1): 4. DOI:10.1186/s13619-020-00046-4 |
| [12] | Colonna Romano N, Fanti L. Transposable elements: major players in shaping genomic and evolutionary patterns. Cells, 2022, 11(6): 1048. DOI:10.3390/cells11061048 |
| [13] | Peterson KM, Mekalanos JJ. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infection and Immunity, 1988, 56(11): 2822-2829. DOI:10.1128/iai.56.11.2822-2829.1988 |
| [14] | Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in Mycobacteria. Proceedings of the National Academy of Sciences of the United States of America, 2001, 98(22): 12712-12717. DOI:10.1073/pnas.231275498 |
| [15] | Van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nature Methods, 2009, 6(10): 767-772. DOI:10.1038/nmeth.1377 |
| [16] | Bai JN, Dai YF, Farinha A, Tang AY, Syal S, Vargas-Cuebas G, Van Opijnen T, Isberg RR, Geisinger E. Essential gene analysis in Acinetobacter baumannii by high-density transposon mutagenesis and CRISPR interference. Journal of Bacteriology, 2021, 203(12): e0056520. |
| [17] | Rifat D, Chen L, Kreiswirth BN, Nuermberger EL. Genome-wide essentiality analysis of Mycobacterium abscessus by saturated transposon mutagenesis and deep sequencing. mBio, 2021, 12(3): e0104921. DOI:10.1128/mBio.01049-21 |
| [18] | Bekaert M, Goffin N, McMillan S, Desbois AP. Essential genes of Vibrio anguillarum and other Vibrio spp. guide the development of new drugs and vaccines. Frontiers in Microbiology, 2021, 12: 755801. DOI:10.3389/fmicb.2021.755801 |
| [19] | Ruiz L, Bottacini F, Boinett CJ, Cain AK, O'Connell-Motherway M, Lawley TD, Van Sinderen D. The essential genomic landscape of the commensal Bifidobacterium breve UCC2003. Scientific Reports, 2017, 7(1): 5648. DOI:10.1038/s41598-017-05795-y |
| [20] | Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathogens, 2011, 7(9): e1002251. DOI:10.1371/journal.ppat.1002251 |
| [21] | Dörr T, Alvarez L, Delgado F, Davis BM, Cava F, Waldor MK. A cell wall damage response mediated by a sensor kinase/response regulator pair enables beta-lactam tolerance. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(2): 404-409. DOI:10.1073/pnas.1520333113 |
| [22] | Karash S, Yahr TL. Genome-wide identification of Pseudomonas aeruginosa genes important for desiccation tolerance on inanimate surfaces. mSystems, 2022, 7(3): e0011422. DOI:10.1128/msystems.00114-22 |
| [23] | Sontheimer EJ. Assembly and function of RNA silencing complexes. Nature Reviews Molecular Cell Biology, 2005, 6(2): 127-138. DOI:10.1038/nrm1568 |
| [24] | Gutbrod MJ, Martienssen RA. Conserved chromosomal functions of RNA interference. Nature Reviews Genetics, 2020, 21(5): 311-331. DOI:10.1038/s41576-019-0203-6 |
| [25] | Liu J, Wang XY, Chen AT, Gao XC, Himes BT, Zhang HY, Chen ZM, Wang JH, Sheu WC, Deng G, Xiao Y, Zou P, Zhang SQ, Liu FY, Zhu Y, Fan R, Patel TR, Saltzman WM, Zhou JB. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nature Communications, 2022, 13(1): 2196. DOI:10.1038/s41467-022-29884-3 |
| [26] | Boyer AP, Collier TS, Vidavsky I, Bose R. Quantitative proteomics with siRNA screening identifies novel mechanisms of trastuzumab resistance in HER2 amplified breast cancers. Molecular & Cellular Proteomics: MCP, 2013, 12(1): 180-193. |
| [27] | Wang L, Zheng J, Luo YZ, Xu T, Zhang QX, Zhang L, Xu MY, Wan JM, Wang MB, Zhang CY, Fan YL. Construction of a genomewide RNAi mutant library in rice. Plant Biotechnology Journal, 2013, 11(8): 997-1005. DOI:10.1111/pbi.12093 |
| [28] | Jin Y, Zhao P, Fang YY, Gao F, Guo HS, Zhao JH. Genome-wide profiling of sRNAs in the Verticillium dahliae-infected Arabidopsis roots. Mycology, 2018, 9(3): 155-165. DOI:10.1080/21501203.2018.1426062 |
| [29] | Cogoni C, Romano N, Macino G. Suppression of gene expression by homologous transgenes. Antonie Van Leeuwenhoek, 1994, 65(3): 205-209. DOI:10.1007/BF00871948 |
| [30] | Nakayashiki H, Nguyen QB. RNA interference: roles in fungal biology. Current Opinion in Microbiology, 2008, 11(6): 494-502. DOI:10.1016/j.mib.2008.10.001 |
| [31] | Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering TL. RNA interference in the pathogenic fungus Cryptococcus neoformans. Genetics, 2002, 160(2): 463-470. DOI:10.1093/genetics/160.2.463 |
| [32] | Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Molecular Cell, 2011, 43(6): 880-891. DOI:10.1016/j.molcel.2011.08.022 |
| [33] | Man S, Cheng RB, Miao CC, Gong QH, Gu YC, Lu XZ, Han F, Yu WG. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Research, 2011, 39(8): e50. DOI:10.1093/nar/gkr034 |
| [34] | Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nature Biotechnology, 2013, 31(2): 170-174. DOI:10.1038/nbt.2461 |
| [35] | Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology, 1987, 169(12): 5429-5433. DOI:10.1128/jb.169.12.5429-5433.1987 |
| [36] | Jansen R, Van Embden JDA, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Molecular Microbiology, 2002, 43(6): 1565-1575. DOI:10.1046/j.1365-2958.2002.02839.x |
| [37] | Horvath P, Romero DA, Coûté-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. Journal of Bacteriology, 2008, 190(4): 1401-1412. DOI:10.1128/JB.01415-07 |
| [38] | Wan F, Draz MS, Gu MJ, Yu W, Ruan Z, Luo QX. Novel strategy to combat antibiotic resistance: a sight into the combination of CRISPR/Cas9 and nanoparticles. Pharmaceutics, 2021, 13(3): 352. DOI:10.3390/pharmaceutics13030352 |
| [39] | Erdmann S, Garrett RA. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Molecular Microbiology, 2012, 85(6): 1044-1056. DOI:10.1111/j.1365-2958.2012.08171.x |
| [40] | Bikard D, Jiang WY, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Research, 2013, 41(15): 7429-7437. DOI:10.1093/nar/gkt520 |
| [41] | Wang XQ, Tokheim C, Gu SS, Wang BB, Tang Q, Li YH, Traugh N, Zeng ZX, Zhang Y, Li ZY, Zhang BN, Fu JX, Xiao TF, Li W, Meyer CA, Chu J, Jiang P, Cejas P, Liu XS. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell, 2021, 184(21): 5357-5374.e22. DOI:10.1016/j.cell.2021.09.006 |
| [42] | Bassaganyas L, Popa SJ, Horlbeck M, Puri C, Stewart SE, Campelo F, Ashok A, Butnaru CM, Brouwers N, Heydari K, Ripoche J, Weissman J, Rubinsztein DC, Schekman R, Malhotra V, Moreau K, Villeneuve J. New factors for protein transport identified by a genome-wide CRISPRi screen in mammalian cells. The Journal of Cell Biology, 2019, 218(11): 3861-3879. DOI:10.1083/jcb.201902028 |
| [43] | Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen YW, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 2014, 159(3): 647-661. DOI:10.1016/j.cell.2014.09.029 |
| [44] | Wang TM, Guan CG, Guo JH, Liu B, Wu YN, Xie Z, Zhang C, Xing XH. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nature Communications, 2018, 9: 2475. DOI:10.1038/s41467-018-04899-x |
| [45] | Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genetics, 2018, 14(11): e1007749. DOI:10.1371/journal.pgen.1007749 |
| [46] | Peters JM, Colavin A, Shi HD, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell, 2016, 165(6): 1493-1506. DOI:10.1016/j.cell.2016.05.003 |
| [47] | Liu X, Gallay C, Kjos M, Domenech A, Slager J, Van Kessel SP, Knoops K, Sorg RA, Zhang JR, Veening JW. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Molecular Systems Biology, 2017, 13(5): 931. DOI:10.15252/msb.20167449 |
| [48] | Liu RM, Liang LY, Freed EF, Choudhury A, Eckert CA, Gill RT. Engineering regulatory networks for complex phenotypes in E. coli. Nature Communications, 2020, 11(1): 4050. DOI:10.1038/s41467-020-17721-4 |
| [49] | Liu J, Liu MS, Shi T, Sun GN, Gao N, Zhao XJ, Guo X, Ni XM, Yuan QQ, Feng JH, Liu ZM, Guo YM, Chen JZ, Wang Y, Zheng P, Sun JB. CRISPR-assisted rational flux-tuning and arrayed CRISPRi screening of an l-proline exporter for l-proline hyperproduction. Nature Communications, 2022, 13(1): 891. DOI:10.1038/s41467-022-28501-7 |
| [50] | Jiang WY, Oikonomou P, Tavazoie S. Comprehensive genome-wide perturbations via CRISPR adaptation reveal complex genetics of antibiotic sensitivity. Cell, 2020, 180(5): 1002-1017.e31. DOI:10.1016/j.cell.2020.02.007 |
| [51] | Huang H, Chai CS, Li N, Rowe P, Minton NP, Yang S, Jiang WH, Gu Y. CRISPR/Cas9-based efficient genome editing in Clostridium ljungdahlii, an autotrophic gas-fermenting bacterium. ACS Synthetic Biology, 2016, 5(12): 1355-1361. DOI:10.1021/acssynbio.6b00044 |
| [52] | Zhao R, Liu YQ, Zhang H, Chai CS, Wang J, Jiang WH, Gu Y. CRISPR-Cas12a-mediated gene deletion and regulation in Clostridium ljungdahlii and its application in carbon flux redirection in synthesis gas fermentation. ACS Synthetic Biology, 2019, 8(10): 2270-2279. DOI:10.1021/acssynbio.9b00033 |
| [53] | Yee T, Furuichi T, Inouye S, Inouye M. Multicopy single-stranded DNA isolated from a Gram-negative bacterium, Myxococcus xanthus. Cell, 1984, 38(1): 203-209. DOI:10.1016/0092-8674(84)90541-5 |
| [54] | Temin HM. Reverse transcriptases retrons in bacteria. Nature, 1989, 339(6222): 254-255. DOI:10.1038/339254a0 |
| [55] | Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, Sorek R. Bacterial retrons function in anti-phage defense. Cell, 2020, 183(6): 1551-1561.e12. DOI:10.1016/j.cell.2020.09.065 |
| [56] | Lopez SC, Crawford KD, Lear SK, Bhattarai-Kline S, Shipman SL. Precise genome editing across kingdoms of life using retron-derived DNA. Nature Chemical Biology, 2022, 18(2): 199-206. DOI:10.1038/s41589-021-00927-y |
| [57] | Schubert MG, Goodman DB, Wannier TM, Kaur D, Farzadfard F, Lu TK, Shipman SL, Church GM. High-throughput functional variant screens via in vivo production of single-stranded DNA. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(18): e2018181118. DOI:10.1073/pnas.2018181118 |