1. 上海理工大学 健康科学与工程学院, 上海 200093;
2. 中科南京生命健康高等研究院, 江苏 南京 211135;
3. 中国科学院上海巴斯德研究所, 上海 200031;
4. 人工智能研究中心 鹏程实验室, 广东 深圳 518055;
5. 上海交通大学 生命科学与生物技术学院, 上海 200240
收稿日期:2021-12-02;接收日期:2022-03-29
基金项目:上海交通大学微生物代谢国家重点实验室开放课题(MMLKF21-11);江苏省创新能力建设计划(BM2020019)
Preparation of luciferase-expressing mRNA and expression characteristics of mRNA delivered by electroporation in vivo
1. School of Health Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China;
2. Nanjing Advanced Academy of Life and Health, Nanjing 211135, Jiangsu, China;
3. Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai 200031, China;
4. Peng Cheng Laboratory, AI Research Center, Shenzhen 518055, Guangdong, China;
5. School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China
Received: December 2, 2021; Accepted: March 29, 2022
Supported by: Open Funding Project of State Key Laboratory of Microbial Metabolism, Shanghai Jiao Tong University (MMLKF21-11); Innovation Capacity Building Project of Jiangsu Province, China (BM2020019)
mRNA目前在疫苗开发、蛋白替代治疗、基因编辑、干细胞重编程和免疫治疗等多个方面均得到了广泛应用[1]。根据mRNA结构元件组成的不同,当前mRNA平台主要分为未修饰的非复制型mRNA、核苷修饰的非复制型mRNA、自复制型的mRNA以及环状mRNA[2]。由于mRNA的不稳定性和高固有免疫原性,mRNA药物的设计需要考虑如何提高mRNA的半衰期、蛋白翻译效率并降低mRNA固有免疫原性,mRNA组成结构的设计优化、mRNA制备纯化以及mRNA的有效递送是mRNA药物开发的主要方面[3-4]。
非复制型mRNA结构简单、片段长度小,其结构元件主要包括开放阅读框(open reading frame, ORF),ORF两侧5′ UTR和3′ UTR两个非翻译区(untranslated region, UTR),一个通过三磷酸酯桥连接到第一个核苷酸上的7-甲基鸟苷(m7G) 并在5′端第一个碱基2位甲基化形成5′的Ⅰ型帽子结构,以及一个3′多聚腺苷酸尾(polyA tail)[3]。5′m7G帽阻断细胞质RNA传感器例如RNA解旋酶视黄酸诱导基因Ⅰ (RIG-Ⅰ) 的识别,抑制5′→3′外切酶介导的降解,招募翻译起始因子,促进高效翻译。5′ UTR和3′ UTR的长度、结构和调控元件的优化均有助于促进蛋白的表达。聚腺苷酸尾及其长度对于翻译和保护mRNA疫苗结构免受降解至关重要[3]。序列工程(密码子优化) 和核苷修饰可以提高翻译效率[4],还会抑制Toll样受体(Toll-like receptor, TLR) 识别和对mRNA结构的先天免疫反应。对于mRNA的制备,保证mRNA纯度至关重要[5]。DNA依赖性RNA聚合酶在RNA合成过程中会产生小的寡核苷酸和双链RNA杂质,去除这些被模式识别受体识别的杂质,通过抑制Ⅰ型干扰素和炎性细胞因子的产生,促进翻译和蛋白质合成[6]。
化学修饰和序列优化改变了蛋白翻译的效率及mRNA的稳定性[4, 7],但裸mRNA作为治疗或免疫手段仍旧存在很大的问题,细胞对mRNA的摄取和转运效率低,因此如何实现mRNA的高效递送是mRNA开发和应用的关键[5-6]。目前mRNA的递送系统可分为物理递送、化学递送和体外树突状细胞(dendritic cells, DCs) 递送[3]。物理递送主要有活体基因导入仪和基因枪两种形式[8],化学递送主要研究的类型包括鱼精蛋白、聚合物、肽类和脂质体等几大类[3, 9]。其中脂质体纳米颗粒(lipid nanoparticle, LNP) 是高效稳定的mRNA递送载体,已成为当前mRNA疫苗开发中最常用的递送系统,但在疫苗开发生产中,一方面其中的核心脂质材料阳离子脂质有专利保护,生产开发成本高;另外,有研究显示LNP靶向肝脏、心脏等器官,可能是肝炎、心肌炎等的诱导因素,其长期的安全性还有待时间的检验[10-11]。电穿孔于1982年首次应用于基因递送,现已成为造血细胞类体外mRNA转染的有效方法,用基因枪进行电穿孔为裸mRNA递送提供了有效途径[12],电穿孔增加了细胞膜的通透性,大大提高了mRNA大分子进入细胞的效率,进而在体内完成目的蛋白的高效表达[12-13]。
为了研究电穿孔介导的mRNA在体内的蛋白表达情况,本研究对编码荧光素酶蛋白的mRNA进行体外转录系统的构建并验证其有效性。体外转录制备的mRNA通过电穿孔递送入小鼠体内,使用小动物活体成像系统通过对荧光光子量的连续观测来判断荧光蛋白表达量及蛋白表达持续时间。
1 材料与方法
1.1 质粒与细胞 质粒pGEM-3Zf(+) 购自Promega公司,HEK293T (人胚胎肾细胞293T) 购自ATCC。
1.2 实验动物 动物实验使用体重16−20 g的雌性7周龄SPF级Balb/c小鼠,购自浙江维通利华实验动物技术有限公司,体内动物实验在上海巴斯德研究所伦理委员会的批准下进行(伦理批号:A2021026)。
1.3 主要试剂和仪器 MEGAscriptTM T7 Transcription Kit、RNase-free Water、Lipofectamine2000购自Thermo fisher公司,Vaccinia Capping System、mRNA Cap 2′-O-methyltransferase、RNase inhibitor购自Novoprotein公司,A-PlusTM Poly(A) Polymerase Tailing Kit、修饰性核苷酸methylpseudouridine-5′-triphosphate购自Cellscript公司,限制性内切酶BamH Ⅰ-HF购自NEB公司,RL6000 RNA marker购自TaKaRa公司,荧光素酶底物d-luciferin购自GLPBIO公司,Luciferase Assay System购自Promega公司,兔源萤火虫荧光素酶单克隆抗体购自Abcam公司,胶回收试剂盒购自QIAGEN公司,LiCl、EDTA、鼠源β-actin单克隆抗体购自Sigma公司。生物安全柜、酶标仪、微量分光光度计NanoDrop 2000购自Thermo fisher公司,IVIS小动物活体成像系统、气体麻醉机购自PerkinElmer公司,冷冻离心机购自GeneSpeed公司,PCR仪购自Bio-Rad公司,基因导入仪购自上海塔瑞莎生物技术有限公司。
1.4 mRNA体外制备 pGEM-3Zf(+) 质粒的T7启动子元件后插入目的mRNA对应的模板序列,包括5′ UTR、ORF和3′ UTR,序列末端引入BamH Ⅰ-HF特异性酶切位点。mRNA体外制备原理示于图 1,即用限制性核酸内切酶BamH Ⅰ-HF对质粒模板进行线性化,通过切胶回收目的片段并使用胶回收试剂盒纯化得到线性化模板DNA。采用转录后加帽加尾的策略体外制备mRNA。以纯化后的线性化质粒为模板,使用MEGAscriptTM T7转录试剂盒按照试剂盒说明书进行体外转录,但用修饰性核苷酸假尿苷m1Ψ-5′-TP代替尿苷酸得到含修饰性核苷酸的RNA产物。使用近岸生物公司的vaccinia capping加帽试剂盒和2′-O-甲基转移酶试剂盒按照标准试剂盒操作流程对RNA进行cap 1加帽。使用A-PlusTM Poly(A) 加尾试剂盒选择试剂盒说明书中的标准加尾体系对mRNA进行加尾,加尾长度约120 nt。每一步RNA产物均使用LiCl (75 mmol/L in EDTA) 沉淀进行纯化,纯化产物使用微量分光光度计(NanoDrop2000) 进行定量,并通过普通琼脂糖凝胶电泳定性,确保结果准确无误后将mRNA最终产物冷冻保存于−80 ℃备用。
1.5 蛋白表达体外验证 为了验证体外转录的mRNA产物的蛋白表达情况,我们使用mRNA转染HEK293T细胞。96孔板设置mRNA转染组和空白对照组,每组两个平行,每孔接种5 000个细胞,使用Lipo2000试剂按照试剂盒说明书进行脂转,每孔转染300 ng mRNA,转染后连续培养24 h后,裂解细胞并使用luciferase相对荧光强度检测试剂盒检测相对荧光活性来间接说明蛋白的表达情况。为了验证体外转录的mRNA产物不同时间的蛋白表达情况,我们使用上述同条件设置mRNA转染组和空白对照组,分别于1、2、5、6、12、24、48、60 h收样并进行检测。同时本研究还使用活体成像仪检测了荧光蛋白表达强度。为了验证蛋白表达,我们还采用了Western blotting法检测,通过收取mRNA转染24 h的293T细胞,裂解后分别用抗β-actin及luciferase抗体进行检测。
1.6 电穿孔介导的mRNA递送 为了进一步验证递送系统的可靠性,以及研究电穿孔介导递送的mRNA于小鼠体内的蛋白表达情况,小鼠随机分成两组,实验组10只,分两笼饲养,每笼5只小鼠,对照组5只,实验组用活体基因导入仪递送裸mRNA,每只小鼠电转量为5 μg mRNA,体积30 μL,基因导入仪选择仪器电转条件为电压30 V,6次脉冲,在小鼠后腿肱四头肌肌肉注射,在注射部位给定短时脉冲导入mRNA;对照组每只小鼠同条件递送30 μL体积的磷酸缓冲盐溶液(phosphate buffered saline, PBS)。
1.7 生物发光成像 小鼠称重,按照150 mg/kg剂量腹腔注射底物d-luciferin (15 mg/mL),小鼠置于预麻盒使用异氟烷进行气体麻醉,经前期底物荧光动力学分析,选择底物注射后第15分钟使用IVIS成像系统进行活体生物发光成像。用Living Image软件v4.4确定目标区域的总光子量。在mRNA导入后4 h和24 h时间点进行小动物活体成像,之后每隔24 h重复活体生物发光成像,直至荧光强度接近背景值停止检测。
1.8 统计分析 使用GraphPad Prism 9.0软件进行统计分析。实验采用单因素方差分析进行分析,使用t检验分析,当P值< 0.05表明差异显著(*: P < 0.05;**: P < 0.01;***: P < 0.001;****: P < 0.000 1)。
2 结果与分析
2.1 构建的体外转录系统 对于mRNA体外制备和储存过程,本研究全部选择使用无酶耗材、无酶试剂并在无RNase污染的洁净环境下进行实验操作,在体外转录、加帽和加尾的制备体系中加入RNase抑制剂(RNase inhibitor),保证在制备过程中RNA不被降解,制备得到的mRNA于–80 ℃保存,保证RNA在长时间储存过程中不被降解;裸mRNA递送至体内后,随时伴随着被RNase降解的风险,一方面RNA在体内可自然降解是RNA应用于药物研发安全性的体现,另一方面需要延长mRNA半衰期保证有效的蛋白表达强度和持续时间,通过非编码区优化、加帽、加尾、修饰性核苷酸的引入以及密码子优化等方式能够抑制RNase对mRNA的识别和降解,从而延长mRNA的半衰期。本研究以荧光素酶蛋白作为指示蛋白,通过小动物活体成像指示了mRNA在小鼠体内表达的持续时间。
构建编码荧光素酶的重组质粒,通过普通琼脂糖凝胶电泳及测序验证质粒序列准确无误,用BamHⅠ-HF进行酶切线性化,各孔重复酶切后的琼脂糖凝胶电泳结果如图 2A,结果显示酶切条带单一、片段大小正确。以线性化的质粒为模板进行体外转录,用LiCl进行纯化,并通过琼脂糖凝胶电泳检验RNA,图 2B中使用已知片段大小的指示用RNA作为对照(control) RNA,结果表明,RNA产物条带单一、大小正确,可用于后续实验。
2.2 mRNA体外转染效果 用mRNA转染HEK293T细胞,验证系统的体外表达情况,转染细胞裂解后检测相对荧光素酶蛋白表达以及相对荧光素酶活性。如图 3A所示,luc-mRNA为转染组,blank为空白对照组,通过检测相同蛋白上样量的不同实验组对应的蛋白表达情况,β-actin作为内参在两组中表达量近一致,空白对照组没有表达出萤火虫荧光素酶蛋白,而转染组明显检测到了萤火虫荧光素酶的蛋白。这表明该系统初步构建成功,可以进一步验证蛋白活性及其功能。图 3B所示,mRNA组为脂转mRNA实验组,control组为脂转未加帽加尾的mRNA对照组、reagent组为脂转试剂对照组、blank组为空细胞对照组,结果表明,mRNA转染组检测到显著(P≤0.001) 高于对照组的荧光强度,即mRNA在HEK293T细胞内成功表达荧光素酶蛋白,进一步说明体外转录系统构建的成功。图 3C表明,荧光素酶蛋白在mRNA转染细胞1 h后即可被检出,在12 h时强烈表达,在24 h之后开始下降,以荧光素酶活性记,从其达到高峰表达的时间点计算,荧光素酶在细胞内的半衰期为12 h。
为了进一步说明转染后细胞内蛋白表达水平,以及体外成像系统的适用性,对mRNA脂转24 h后的HEK293T细胞进行体外成像,如图 3D所示,luc-mRNA为脂转mRNA实验组,并设置300 ng/孔mRNA转染的实验组(蓝色方框内孔,孔B2、B3、B5、B7;每孔转染600 ng/μL mRNA),转染mRNA但未加底物对照组(黄色方框内孔,孔B10、B11;每孔转染600 ng/μL mRNA),control为脂转未加帽加尾的RNA对照组(红色方框内孔,孔G10、G11)。图 3D为经IVIS成像系统的分析软件living image对成像系统捕获的发射光进行定量和显示得到的伪彩图,不同的颜色代表了不同的发光值(光子数),由蓝色到红色的渐变分别对应发光值由低到高的变化,右侧的颜色列表展示了1.0×107−5.0×107 p/(s·cm2·sr) 不同发光值(光子数) 对应的颜色。发光值间接反映荧光素酶蛋白的表达量。结果表明,对照组未检测到荧光强度,300 ng/孔转染量的实验组检测到较高强度的荧光,对应较高的蛋白表达水平,成像系统适用于所构建的mRNA体外转录系统,可进一步用于小鼠体内转染效果评价。
2.3 mRNA体内转导效果
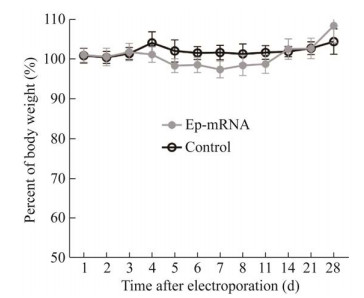
2.3.1 实验动物观察 对电导后的小鼠进行连续观测发现,实验组(Ep-mRNA) 和对照组(control) 小鼠皮毛颜色正常,状态良好,处于健康的生命状态,连续观测的两周内体重结果(图 4) 表明,小鼠体重在电转mRNA前后无明显差异,电导mRNA未对小鼠产生明显的副作用。
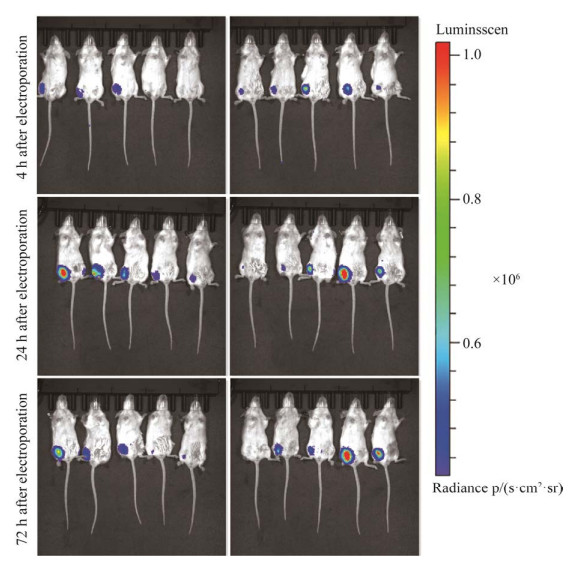
2.3.2 mRNA体内蛋白表达 为了研究电穿孔介导的mRNA在小鼠体内的蛋白表达情况,肌肉电穿孔给药后使用小动物活体成像系统连续监测蛋白表达,监测周期为28 d。生物发光图像可以观测到蛋白表达部位及蛋白表达强度的分布,图 5为经IVIS成像系统的分析软件living image对成像系统捕获的发射光进行定量和显示得到的伪彩图,不同的颜色代表了不同的发光值,由蓝色到红色的渐变分别对应发光值由低到高的变化,右侧颜色列表展示了0.4×106–1.0×106 p/(s·cm2·sr) 不同发光值对应的颜色。mRNA电导后4 h,8只小鼠在注射部位观测到荧光表达,两只未观测到;电导后24 h,10只小鼠均观测到荧光强度,包括之前未检测到的两只,其中有两只小鼠荧光强度较第4小时呈降低趋势,其余小鼠荧光强度均呈增强趋势;电导后72 h,有1只小鼠未观测到荧光,荧光强度整体较第24小时呈下降趋势,但有3只小鼠观测到的荧光强度仍维持或呈增强趋势。通过小动物活体成像中ROI值的计算可以更直观地显示各个小鼠荧光素酶作用于底物产生的荧光情况,图 6则展示了各个小鼠电导mRNA后检测到的总光子通量,由图 6可知小鼠个体间蛋白表达差异明显,尤其在mRNA转导后的第1到第4天,总光子通量达到一个数量级的差异;在第48小时,实验组小鼠整体的总光子通量达到峰值;在第14天,所有小鼠总光子通量基本达到一致。
所有小鼠均成功表达了荧光素酶蛋白,光子总量最大值达到了107/s,表明电穿孔介导的mRNA的体内表达是成功的,但蛋白表达强度和持续时间存在较大的小鼠个体间差异。
3 讨论 荧光素酶是常用的蛋白表达指示蛋白[6],可以作用于荧光素酶底物产生荧光,荧光强度与荧光素酶表达量呈正相关,相较于生物发光法,荧光法检测灵敏度高且能够很好地降低小鼠活体成像的背景噪音,故本研究选择萤火虫荧光素酶为靶标蛋白研究非复制型mRNA的构建和蛋白体内表达。
非复制型mRNA的平台构建包括mRNA序列设计、mRNA的制备和mRNA的递送和检测3个部分[3]。mRNA的组成结构元件及整体序列结构的设计和改造对于提高mRNA的稳定性和翻译效率至关重要,是mRNA应用平台的核心技术之一[3, 5]。对非编码区的优化可有效提高mRNA的稳定性,5′ UTR还对mRNA翻译效率的提高具有重要影响,引入经典Kozak序列提高翻译效率和避免稳定二级结构的出现是该序列区域两个重要关注点[7, 4-15]。目前有多种来源和改造的UTR可选择,但是尚未有最佳选择的标准依据。当前人β-珠蛋白和α-珠蛋白的非编码区序列选用最为广泛,也是本研究中选择使用的序列[5, 15]。对编码区密码子的优化可以有效提高蛋白表达量,随着人们对密码子和蛋白翻译过程的研究深入,密码子优化的概念和策略也在不断细化和推进,以达到提高蛋白翻译速率和保证蛋白折叠准确性相平衡的目的[7, 15],本研究使用成熟的密码子优化软件对荧光素酶编码基因进行了鼠源密码子优化。帽子结构是真核细胞翻译起始识别位点,也可保护其不被RNA核酸外切酶识别,延缓mRNA降解速率,是mRNA体外构建必不可少的一部分[3, 12, 16]。真核细胞内mRNA的降解通常是从聚腺苷酸尾(polyA) 的降解开始的,除此之外polyA也参与mRNA的翻译起始,有研究显示,polyA尾长度为120 nt可以达到最好的蛋白翻译效率[15, 17],但在不同细胞种类中显示出差异,本研究选择120 nt的加尾长度。对于整体序列,还需关注GC含量、过度稳定的mRNA二级结构和小RNA结合位点等[3, 9, 15]。
使用修饰性核苷酸可以显著降低mRNA的固有免疫原性,从而进一步提高mRNA的半衰期,降低mRNA副作用[4, 18]。该平台选择使用修饰性核苷酸7-甲基-假尿嘧啶代替尿嘧啶(U)。对于mRNA的制备,体外转录模板可以选择PCR扩增法或质粒线性化两种方式制备,考虑到突变位点引入概率我们选择线性化质粒为模板,并用转录后加帽加尾的策略制备mRNA。用高效转录的T7 RNA聚合酶进行体外转录,使用酶法进行两步加帽可以达到99%以上的Ⅰ型帽子结构的加帽效率,酶法加尾可通过调整加尾体系和孵育时间等条件得到适宜的加尾长度[19]。T7 RNA聚合酶转录过程中会产生双链RNA (double-stranded RNA, dsRNA),用高效液相色谱法或纤维素法对mRNA进行纯化可以有效降低dsRNA的污染,从而进一步降低mRNA的固有免疫原性和提高mRNA翻译效率[20]。考虑到dsRNA的含量较低且有研究显示固有免疫原性的降低可能是mRNA二级结构被打开,该研究中mRNA的制备暂未做进一步纯化处理。按照以上序列设计和制备方法得到的mRNA样品,经体外转染HEK293T检测蛋白表达,初步验证了该表达系统的可行性且蛋白获得表达。
mRNA为带负电的大分子,需要借助物理或化学方法才能达到有效的胞内递送[18, 21]。电转通过改变细胞膜通透性使得mRNA大分子易于穿过细胞膜进入细胞质,之后细胞膜恢复初始状态[14, 16]。研究表明,脂质体递送可以有效保护mRNA不被核酸酶快速降解,延长mRNA半衰期,提高mRNA的细胞摄取效率[11, 22]。本研究选择电穿孔方式进行mRNA的体内递送,一方面电穿孔技术已广泛应用于DNA等大分子的递送,具有一定的安全性和有效性保障,另一方面可以避开其他外源化学物质的引入,排除对研究mRNA表达的干扰。
外源mRNA会被细胞内的固有免疫识别受体例如Toll样受体(TLR) 识别,诱导产生炎症反应[23];电转也可能造成一定的物理损伤[14, 24],所以mRNA经电穿孔介导进入小鼠体内后,我们对小鼠的健康状况进行了监测并重点关注了小鼠体重指标,以确定该mRNA平台的安全性,结果表明,电穿孔的方式未对小鼠体重造成显著性下降,健康状况也未受到影响。
靶标蛋白的表达强度和持续时间是mRNA药物评价重要的指标之一[25-26]。本研究通过小动物活体成像系统检测荧光素酶作用于底物产生的荧光强度间接反映荧光素酶蛋白的表达量和持续时间,结果显示本研究实现了mRNA在小鼠体内的原位表达。实验中观测到小鼠个体间蛋白表达强度和蛋白活性持续时间不一致的情况,造成该差异的可能原因包括小鼠个体间差异、mRNA的不稳定性、基因导入仪仪器本身的不稳定性以及实验操作差异例如注射部位的选择、电极插入深度等,对于该不一致性的情况仍需在未来进行深入探究。
本研究构建了一种电穿孔介导的非复制型mRNA平台,研究了mRNA在小鼠体内的表达特征。该平台可在非编码区优化、密码子优化、mRNA纯化、mRNA递送等方面进行进一步的优化并有望应用于mRNA疫苗或基因治疗药物的研发。
 2022, Vol. 38
2022, Vol. 38